
Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. DNA
3. Results
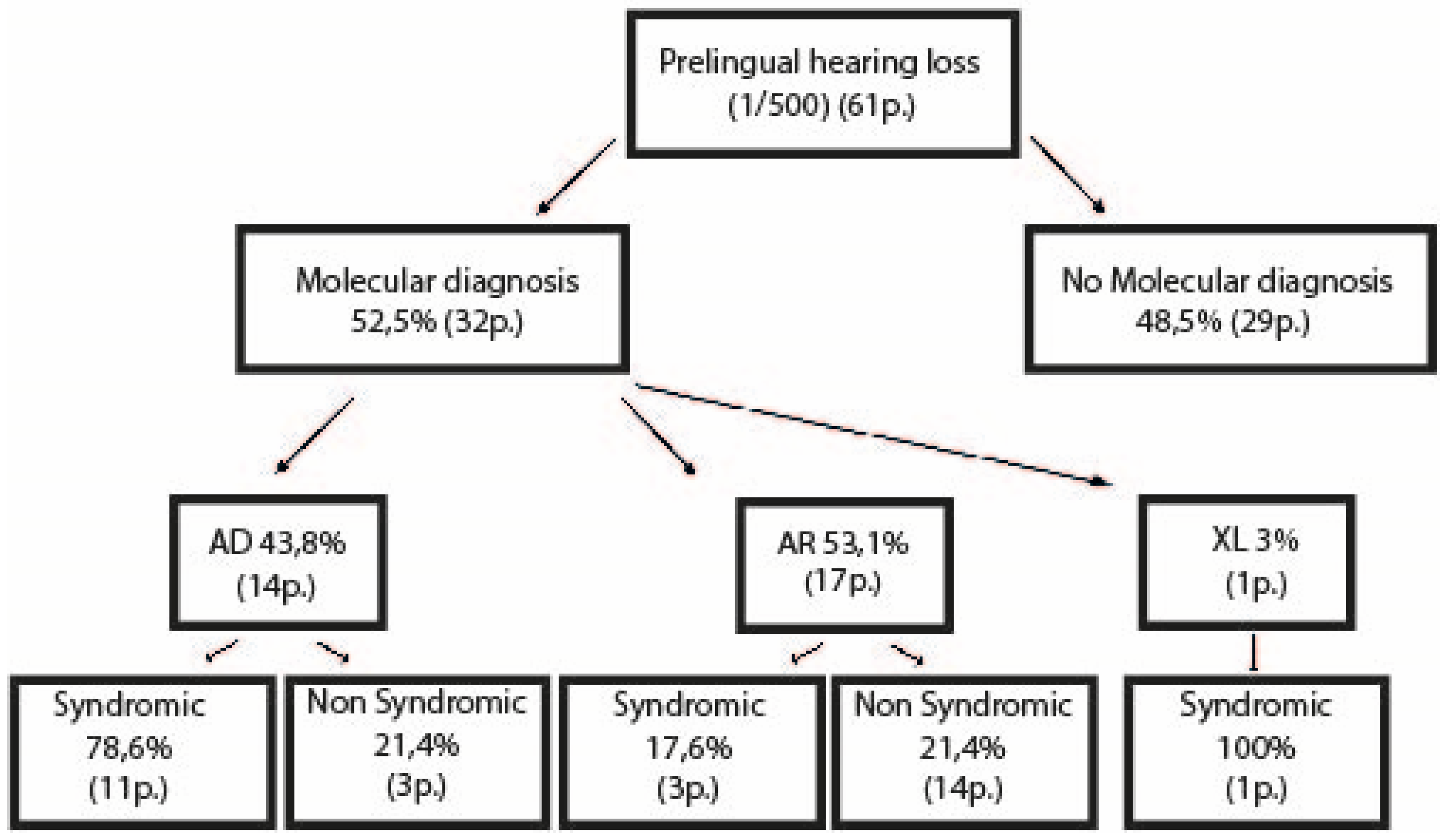
3.1. Cohort Descriptions
3.1.1. Children
3.1.2. Adults
3.1.3. Patients Identified through Direct Sequencing of GJB2/GJB6
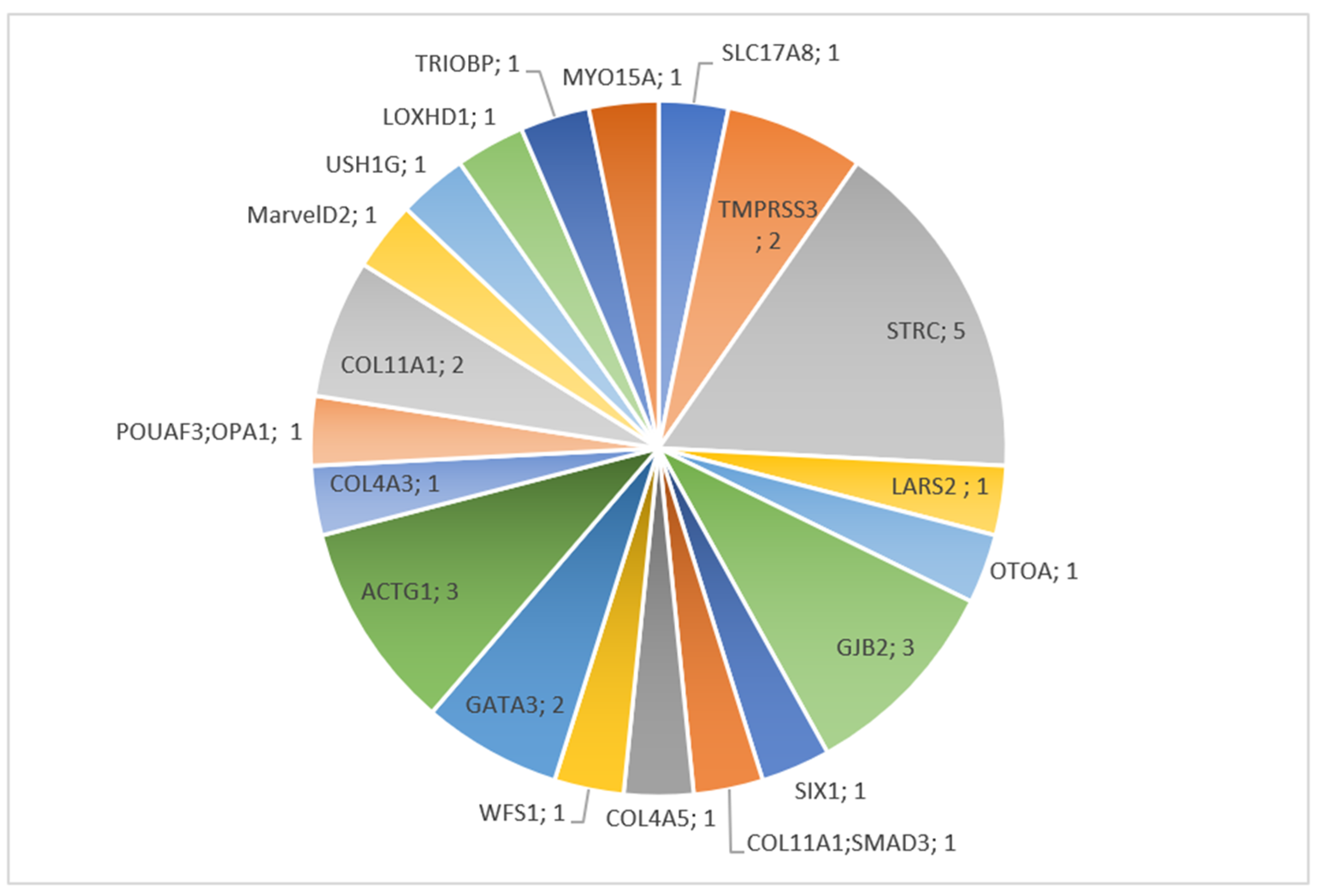
3.2. Molecular Results
3.2.1. Children
3.2.2. Adults
3.3. Brief Description of Individual Cases Confirmed by Molecular Diagnosis
3.3.1. COL4A5
3.3.2. USH1G
3.3.3. GJB2
3.3.4. SIX1
3.3.5. LARS2
3.3.6. ILDR1
3.3.7. ACTG1
3.3.8. GATA3
3.3.9. SLC17A8
3.3.10. LOXHD1
3.3.11. OTOA
3.3.12. WSF1
3.3.13. STRC
3.3.14. POU4F3 and OPA1
3.3.15. COL11A1
3.3.16. COL11A1 and SMAD3
3.3.17. TRIOBP
3.3.18. TMPRSS3
3.3.19. COL4A3
3.3.20. MARVELD2
3.3.21. MYO15A
3.3.22. NF2
3.3.23. COCH
3.4. Variant/s of Unknown Significance (VUS)
3.5. Molecular Results through Direct Sequencing of GJB2-GJB6
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BTE | behind-the-ear |
| CNVs | copy number variant/s |
| CT | computed tomography |
| ENT | ear-nose-throat |
| HL | hearing loss |
| MLPA | multiplex ligation-dependent probe amplification |
| MRI | magnetic resonance imaging |
| OEA | otoacoustic emissions |
| SN | sensorineural |
| VUS | variant of unknown significance |
| WES | whole-exome sequencing |
References
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef]
- Morton, N.E. Genetic epidemiology of hearing impairment. Ann. N. Y. Acad. Sci. 1991, 630, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Jilla, A.M.; Reed, N.S.; Oh, E.S.; Lin, F.R. A Geriatrician’s Guide to Hearing Loss. J. Am. Geriatr. Soc. 2021, 69, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Marazita, M.L.; Ploughman, L.M.; Rawlings, B.; Remington, E.; Arnos, K.S.; Nance, W.E. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am. J. Med. Genet. 1993, 46, 486–491. [Google Scholar] [CrossRef]
- Smith, R.J.; Bale, J.F., Jr.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Nelson, H.D.; Bougatsos, C.; Nygren, P.; 2001 US Preventive Services Task Force. Universal newborn hearing screening: Systematic review to update the 2001 US Preventive Services Task Force Recommendation. Pediatrics 2008, 122, e266–e276. [Google Scholar] [CrossRef]
- Grandori, F. The European Consensus Development Conference on Neonatal Hearing Screening (Milan, 15–16 May 1998). Arch. Otolaryngol. Head Neck Surg. 1999, 125, 118. [Google Scholar] [CrossRef]
- Liming, B.J.; Carter, J.; Cheng, A.; Choo, D.; Curotta, J.; Carvalho, D.; Germiller, J.A.; Hone, S.; Kenna, M.A.; Loundon, N.; et al. International Pediatric Otolaryngology Group (IPOG) consensus recommendations: Hearing loss in the pediatric patient. Int. J. Pediatr. Otorhinolaryngol. 2016, 90, 251–258. [Google Scholar] [CrossRef]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J.H. Hereditary Hearing Loss and Deafness Overview; GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Smith, R.H.; Van Camp, G. Deafness and Hereditary Hearing Loss Overview; GeneReviews®; University of Washington: Seattle, WA, USA, 2010. [Google Scholar]
- Ahmadmehrabi, S.; Brant, J.; Epstein, D.J.; Ruckenstein, M.J.; Rader, D.J. Genetics of Postlingual Sensorineural Hearing Loss. Laryngoscope 2021, 131, 401–409. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joInt. consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Project, N.E.S.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef]
- Fromer, M.; Purcell, S.M. Using XHMM Software to Detect Copy Number Variation in Whole-Exome Sequencing Data. Curr. Protoc. Hum. Genet. 2014, 81, 7.23.1–7.23.21. [Google Scholar] [CrossRef] [PubMed]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Savige, J.; Colville, D.; Rheault, M.; Gear, S.; Lennon, R.; Lagas, S.; Finlay, M.; Flinter, F. Alport Syndrome in Women and Girls. Clin. J. Am. Soc. Nephrol. 2016, 11, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.F.; Hostikka, S.L.; Zhou, J.; Chow, L.T.; Oliphant, A.R.; Gerken, S.C.; Gregory, M.C.; Skolnick, M.H.; Atkin, C.L.; Tryggvason, K. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 1990, 248, 1224–1227. [Google Scholar] [CrossRef]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 2003, 14, 2603–2610. [Google Scholar] [CrossRef]
- Pennings, R.J.; Huygen, P.L.; Orten, D.J.; Wagenaar, M.; van Aarem, A.; Kremer, H.; Kimberling, W.J.; Cremers, C.W.; Deutman, A.F. Evaluation of visual impairment in Usher syndrome 1b and Usher syndrome 2a. Acta Ophthalmol. Scand. 2004, 82, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; White, T.W.; Smith, L.E.; Bailey, R.A.; Compton, J.G.; Paul, D.L.; Bale, S.J. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum. Genet. 1998, 103, 393–399. [Google Scholar] [CrossRef]
- Uyguner, O.; Tukel, T.; Baykal, C.; Eris, H.; Emiroglu, M.; Hafiz, G.; Ghanbari, A.; Baserer, N.; Yuksel-Apak, M.; Wollnik, B. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clin. Genet. 2002, 62, 306–309. [Google Scholar] [CrossRef]
- Ruf, R.G.; Xu, P.X.; Silvius, D.; Otto, E.A.; Beekmann, F.; Muerb, U.T.; Kumar, S.; Neuhaus, T.J.; Kemper, M.J.; Raymond, R.M., Jr.; et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc. Natl. Acad. Sci. USA 2004, 101, 8090–8095. [Google Scholar] [CrossRef]
- Carminho-Rodrigues, M.T.; Klee, P.; Laurent, S.; Guipponi, M.; Abramowicz, M.; Cao-van, H.; Guinand, N.; Paoloni-Giacobino, A. LARS2-Perrault syndrome: A new case report and literature review. BMC Med. Genet. 2020, 21, 109. [Google Scholar] [CrossRef]
- Borck, G.; Ur Rehman, A.; Lee, K.; Pogoda, H.M.; Kakar, N.; von Ameln, S.; Grillet, N.; Hildebrand, M.S.; Ahmed, Z.M.; Nurnberg, G.; et al. Loss-of-function mutations of ILDR1 cause autosomal-recessive hearing impairment DFNB42. Am. J. Hum. Genet. 2011, 88, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Kemerley, A.; Sloan, C.; Pfeifer, W.; Smith, R.; Drack, A. A novel mutation in ACTG1 causing Baraitser-Winter syndrome with extremely variable expressivity in three generations. Ophthalmic Genet. 2017, 38, 152–156. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, X.; Huang, B.; Lu, J.; Wang, G.; Lin, X.; Qu, Y.; Dai, P. Phenotypic Heterogeneity in a DFNA20/26 family segregating a novel ACTG1 mutation. BMC Genet. 2016, 17, 33. [Google Scholar] [CrossRef]
- Miyajima, H.; Moteki, H.; Day, T.; Nishio, S.Y.; Murata, T.; Ikezono, T.; Takeda, H.; Abe, S.; Iwasaki, S.; Takahashi, M.; et al. Novel ACTG1 mutations in patients identified by massively parallel DNA sequencing cause progressive hearing loss. Sci. Rep. 2020, 10, 7056. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Groenen, P.; Nesbit, M.A.; Schuffenhauer, S.; Lichtner, P.; Vanderlinden, G.; Harding, B.; Beetz, R.; Bilous, R.W.; Holdaway, I.; et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature 2000, 406, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Thirlwall, A.S.; Brown, D.J.; McMillan, P.M.; Barker, S.E.; Lesperance, M.M. Phenotypic characterization of hereditary hearing impairment linked to DFNA25. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 830–835. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riazuddin, S.A.; Parker, D.S.; McGlumphy, E.J.; Oh, E.C.; Iliff, B.W.; Schmedt, T.; Jurkunas, U.; Schleif, R.; Katsanis, N.; Gottsch, J.D. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am. J. Hum. Genet. 2012, 90, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Smith, R.J. Massively Parallel Sequencing for Genetic Diagnosis of Hearing Loss: The New Standard of Care. Otolaryngol. Head Neck Surg. 2015, 153, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Gehrig, C.; Nouspikel, T.; Amr, S.S.; Oza, A.; Murphy, E.; Vannier, A.; Bena, F.S.; Carminho-Rodrigues, M.T.; Blouin, J.L.; et al. Molecular characterization of pathogenic OTOA gene conversions in hearing loss patients. Hum. Mutat. 2021, 42, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Chiu, I.; Santos-Cortez, R.L.; Basit, S.; Khan, S.; Azeem, Z.; Andrade, P.B.; Kim, S.S.; Ahmad, W.; Leal, S.M. Novel OTOA mutations cause autosomal recessive non-syndromic hearing impairment in Pakistani families. Clin. Genet. 2013, 84, 294–296. [Google Scholar] [CrossRef]
- Valero, R.; Bannwarth, S.; Roman, S.; Paquis-Flucklinger, V.; Vialettes, B. Autosomal dominant transmission of diabetes and congenital hearing impairment secondary to a missense mutation in the WFS1 gene. Diabet. Med. 2008, 25, 657–661. [Google Scholar] [CrossRef]
- Vona, B.; Hofrichter, M.A.; Neuner, C.; Schroder, J.; Gehrig, A.; Hennermann, J.B.; Kraus, F.; Shehata-Dieler, W.; Klopocki, E.; Nanda, I.; et al. DFNB16 is a frequent cause of congenital hearing impairment: Implementation of STRC mutation analysis in routine diagnostics. Clin. Genet. 2015, 87, 49–55. [Google Scholar] [CrossRef]
- Kim, H.J.; Won, H.H.; Park, K.J.; Hong, S.H.; Ki, C.S.; Cho, S.S.; Venselaar, H.; Vriend, G.; Kim, J.W. SNP linkage analysis and whole exome sequencing identify a novel POU4F3 mutation in autosomal dominant late-onset nonsyndromic hearing loss (DFNA15). PLoS ONE 2013, 8, e79063. [Google Scholar] [CrossRef]
- Pesch, U.E.; Leo-Kottler, B.; Mayer, S.; Jurklies, B.; Kellner, U.; Apfelstedt-Sylla, E.; Zrenner, E.; Alexander, C.; Wissinger, B. OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum. Mol. Genet. 2001, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Ham, M.; Han, J.; Osann, K.; Smith, M.; Kimonis, V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion 2019, 46, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef]
- Boothe, M.; Morris, R.; Robin, N. Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation. J. Pers Med. 2020, 10, 105. [Google Scholar] [CrossRef]
- Rad, A.; Schade-Mann, T.; Gamerdinger, P.; Yanus, G.A.; Schulte, B.; Muller, M.; Imyanitov, E.N.; Biskup, S.; Lowenheim, H.; Tropitzsch, A.; et al. Aberrant COL11A1 splicing causes prelingual autosomal dominant nonsyndromic hearing loss in the DFNA37 locus. Hum. Mutat. 2021, 42, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef]
- Riazuddin, S.; Khan, S.N.; Ahmed, Z.M.; Ghosh, M.; Caution, K.; Nazli, S.; Kabra, M.; Zafar, A.U.; Chen, K.; Naz, S.; et al. Mutations in TRIOBP, which encodes a putative cytoskeletal-organizing protein, are associated with nonsyndromic recessive deafness. Am. J. Hum. Genet. 2006, 78, 137–143. [Google Scholar] [CrossRef]
- Wattenhofer, M.; Di Iorio, M.V.; Rabionet, R.; Dougherty, L.; Pampanos, A.; Schwede, T.; Montserrat-Sentis, B.; Arbones, M.L.; Iliades, T.; Pasquadibisceglie, A.; et al. Mutations in the TMPRSS3 gene are a rare cause of childhood nonsyndromic deafness in Caucasian patients. J. Mol. Med. 2002, 80, 124–131. [Google Scholar] [CrossRef]
- Rosado, C.; Bueno, E.; Fraile, P.; Garcia-Cosmes, P.; Gonzalez-Sarmiento, R. A new mutation in the COL4A3 gene responsible for autosomal dominant Alport syndrome, which only generates hearing loss in some carriers. Eur. J. Med. Genet. 2015, 58, 35–38. [Google Scholar] [CrossRef]
- Nayak, G.; Varga, L.; Trincot, C.; Shahzad, M.; Friedman, P.L.; Klimes, I.; Greinwald, J.H., Jr.; Riazuddin, S.A.; Masindova, I.; Profant, M.; et al. Molecular genetics of MARVELD2 and clinical phenotype in Pakistani and Slovak families segregating DFNB49 hearing loss. Hum. Genet. 2015, 134, 423–437. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Guan, J.; Wang, H.; Yin, L.; Wang, D.; Zhao, L.; Zhou, H.; Wang, Q. Genotype-phenotype correlation analysis of MYO15A variants in autosomal recessive non-syndromic hearing loss. BMC Med. Genet. 2019, 20, 60. [Google Scholar] [CrossRef] [PubMed]
- Kresak, J.L.; Walsh, M. Neurofibromatosis: A Review of NF1, NF2, and Schwannomatosis. J. Pediatr. Genet. 2016, 5, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Kemperman, M.H.; Bom, S.J.; Lemaire, F.X.; Verhagen, W.I.; Huygen, P.L.; Cremers, C.W. DFNA9/COCH and its phenotype. Adv. Otorhinolaryngol. 2002, 61, 66–72. [Google Scholar] [CrossRef]
- Chakchouk, I.; Grati, M.; Bademci, G.; Bensaid, M.; Ma, Q.; Chakroun, A.; Foster, J., 2nd; Yan, D.; Duman, D.; Diaz-Horta, O.; et al. Novel mutations confirm that COL11A2 is responsible for autosomal recessive non-syndromic hearing loss DFNB53. Mol. Genet. Genom. 2015, 290, 1327–1334. [Google Scholar] [CrossRef]
- Balestrini, S.; Milh, M.; Castiglioni, C.; Luthy, K.; Finelli, M.J.; Verstreken, P.; Cardon, A.; Strazisar, B.G.; Holder, J.L., Jr.; Lesca, G.; et al. TBC1D24 genotype-phenotype correlation: Epilepsies and other neurologic features. Neurology 2016, 87, 77–85. [Google Scholar] [CrossRef]
- Azaiez, H.; Booth, K.T.; Bu, F.; Huygen, P.; Shibata, S.B.; Shearer, A.E.; Kolbe, D.; Meyer, N.; Black-Ziegelbein, E.A.; Smith, R.J. TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum. Mutat. 2014, 35, 819–823. [Google Scholar] [CrossRef]
- Astuto, L.M.; Bork, J.M.; Weston, M.D.; Askew, J.W.; Fields, R.R.; Orten, D.J.; Ohliger, S.J.; Riazuddin, S.; Morell, R.J.; Khan, S.; et al. CDH23 mutation and phenotype heterogeneity: A profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am. J. Hum. Genet. 2002, 71, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Jouret, G.; Poirsier, C.; Spodenkiewicz, M.; Jaquin, C.; Gouy, E.; Arndt, C.; Labrousse, M.; Gaillard, D.; Doco-Fenzy, M.; Lebre, A.S. Genetics of Usher Syndrome: New Insights From a Meta-analysis. Otol. Neurotol. 2019, 40, 121–129. [Google Scholar] [CrossRef]
- Ahmed, Z.M.; Riazuddin, S.; Aye, S.; Ali, R.A.; Venselaar, H.; Anwar, S.; Belyantseva, P.P.; Qasim, M.; Riazuddin, S.; Friedman, T.B. Gene structure and mutant alleles of PCDH15: Nonsyndromic deafness DFNB23 and type 1 Usher syndrome. Hum. Genet. 2008, 124, 215–223. [Google Scholar] [CrossRef]
- Toualbi, L.; Toms, M.; Moosajee, M. USH2A-retinopathy: From genetics to therapeutics. Exp. Eye Res. 2020, 201, 108330. [Google Scholar] [CrossRef]
- Thoenes, M.; Zimmermann, U.; Ebermann, I.; Ptok, M.; Lewis, M.A.; Thiele, H.; Morlot, S.; Hess, M.M.; Gal, A.; Eisenberger, T.; et al. OSBPL2 encodes a protein of inner and outer hair cell stereocilia and is mutated in autosomal dominant hearing loss (DFNA67). Orphanet J. Rare Dis. 2015, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Sommen, M.; Wuyts, W.; Van Camp, G. Molecular diagnostics for hereditary hearing loss in children. Expert Rev. Mol. Diagn. 2017, 17, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Downie, L.; Amor, D.J.; Halliday, J.; Lewis, S.; Martyn, M.; Goranitis, I. Exome Sequencing for Isolated Congenital Hearing Loss: A Cost-Effectiveness Analysis. Laryngoscope 2021, 131, E2371–E2377. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinform. 2013, 14 (Suppl. 11), S1. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, N.K.; Kim, A.R.; Rhee, J.; Oh, S.H.; Koo, J.W.; Nam, J.Y.; Park, W.Y.; Choi, B.Y. Exploration of molecular genetic etiology for Korean cochlear implantees with severe to profound hearing loss and its implication. Orphanet J. Rare Dis. 2014, 9, 167. [Google Scholar] [CrossRef]
- Chen, S.; Dong, C.; Wang, Q.; Zhong, Z.; Qi, Y.; Ke, X.; Liu, Y. Targeted Next-Generation Sequencing Successfully Detects Causative Genes in Chinese Patients with Hereditary Hearing Loss. Genet. Test. Mol. Biomark. 2016, 20, 660–665. [Google Scholar] [CrossRef]
- Iwasa, Y.I.; Nishio, S.Y.; Usami, S.I. Comprehensive Genetic Analysis of Japanese Autosomal Dominant Sensorineural Hearing Loss Patients. PLoS ONE 2016, 11, e0166781. [Google Scholar] [CrossRef]
- Christensen, K.; Frederiksen, H.; Hoffman, H.J. Genetic and environmental influences on self-reported reduced hearing in the old and oldest old. J. Am. Geriatr. Soc. 2001, 49, 1512–1517. [Google Scholar] [CrossRef]
- Lewis, M.A.; Nolan, L.S.; Cadge, B.A.; Matthews, L.J.; Schulte, B.A.; Dubno, J.R.; Steel, K.P.; Dawson, S.J. Whole exome sequencing in adult-onset hearing loss reveals a high load of predicted pathogenic variants in known deafness-associated genes and identifies new candidate genes. BMC Med. Genom. 2018, 11, 77. [Google Scholar] [CrossRef]
- Zazo Seco, C.; Wesdorp, M.; Feenstra, I.; Pfundt, R.; Hehir-Kwa, J.Y.; Lelieveld, S.H.; Castelein, S.; Gilissen, C.; de Wijs, I.J.; Admiraal, R.J.; et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur. J. Hum. Genet. 2017, 25, 308–314. [Google Scholar] [CrossRef]
- Morin, M.; Bryan, K.E.; Mayo-Merino, F.; Goodyear, R.; Mencia, A.; Modamio-Hoybjor, S.; del Castillo, I.; Cabalka, J.M.; Richardson, G.; Moreno, F.; et al. In vivo and in vitro effects of two novel γ-actin (ACTG1) mutations that cause DFNA20/26 hearing impairment. Hum. Mol. Genet. 2009, 18, 3075–3089. [Google Scholar] [CrossRef]
- Battelino, S.; Klancar, G.; Kovac, J.; Battelino, T.; Trebusak Podkrajsek, K. TMPRSS3 mutations in autosomal recessive nonsyndromic hearing loss. Eur. Arch. Otorhinolaryngol. 2016, 273, 1151–1154. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef]
- Rudman, J.R.; Kabahuma, R.I.; Bressler, S.E.; Feng, Y.; Blanton, S.H.; Yan, D.; Liu, X.Z. The genetic basis of deafness in populations of African descent. J. Genet. Genom. 2017, 44, 285–294. [Google Scholar] [CrossRef]
- Wu, C.C.; Tsai, C.Y.; Lin, Y.H.; Chen, P.Y.; Lin, P.H.; Cheng, Y.F.; Wu, C.M.; Lin, Y.H.; Lee, C.Y.; Erdenechuluun, J.; et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772. [Google Scholar] [CrossRef]
- Smith, E.D.; Blanco, K.; Sajan, S.A.; Hunter, J.M.; Shinde, D.N.; Wayburn, B.; Rossi, M.; Huang, J.; Stevens, C.A.; Muss, C.; et al. A retrospective review of multiple findings in diagnostic exome sequencing: Half are distinct and half are overlapping diagnoses. Genet. Med. 2019, 21, 2199–2207. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Heurck, R.; Carminho-Rodrigues, M.T.; Ranza, E.; Stafuzza, C.; Quteineh, L.; Gehrig, C.; Hammar, E.; Guipponi, M.; Abramowicz, M.; Senn, P.; et al. Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss. Genes 2021, 12, 1277. https://doi.org/10.3390/genes12081277
Van Heurck R, Carminho-Rodrigues MT, Ranza E, Stafuzza C, Quteineh L, Gehrig C, Hammar E, Guipponi M, Abramowicz M, Senn P, et al. Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss. Genes. 2021; 12(8):1277. https://doi.org/10.3390/genes12081277
Chicago/Turabian StyleVan Heurck, Roxane, Maria Teresa Carminho-Rodrigues, Emmanuelle Ranza, Caterina Stafuzza, Lina Quteineh, Corinne Gehrig, Eva Hammar, Michel Guipponi, Marc Abramowicz, Pascal Senn, and et al. 2021. "Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss" Genes 12, no. 8: 1277. https://doi.org/10.3390/genes12081277
APA StyleVan Heurck, R., Carminho-Rodrigues, M. T., Ranza, E., Stafuzza, C., Quteineh, L., Gehrig, C., Hammar, E., Guipponi, M., Abramowicz, M., Senn, P., Guinand, N., Cao-Van, H., & Paoloni-Giacobino, A. (2021). Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss. Genes, 12(8), 1277. https://doi.org/10.3390/genes12081277