
Genetic Evaluation of Inpatient Neonatal and Infantile Congenital Heart Defects: New Findings and Review of the Literature



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Inclusion Criteria
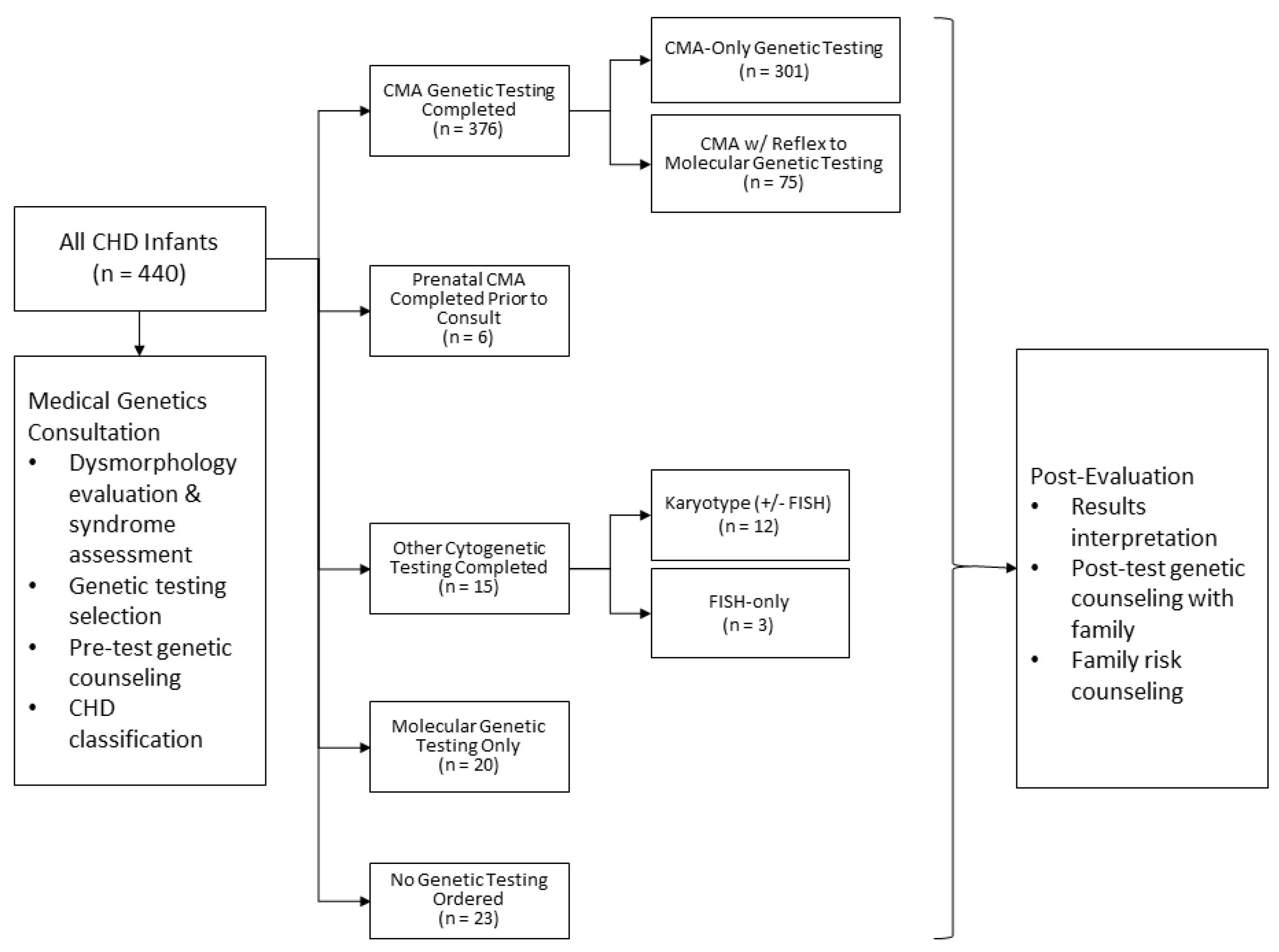
2.2. Clinical Algorithm for Inpatient Cardiovascular Genetics Evaluations
2.3. CHD Classification
2.4. Clinical Evaluations and Defining Apparently Isolated vs. Syndromic CHDs
2.5. Genetic Testing and Results Classification
2.6. Descriptive Statistics and Analyses
3. Results
3.1. Overview of Cohort
3.2. Overview of Genetic Testing Practices
3.3. Diagnostic Yield of CMA
3.4. Geneticists’ a Priori Assessment of Likelihood of Genetic Testing Abnormalities
3.4.1. APVR
3.4.2. AVSD
3.4.3. Complex
3.4.4. Conotruncal
3.4.5. Heterotaxy
3.4.6. LVOTO
3.4.7. RVOTO
3.4.8. Septal
3.5. Results of Molecular Testing
4. Discussion
4.1. CHD Type Influences Diagnostic Yield
4.2. Dysmorphic Features and/or ECAs Are Associated with Increased Diagnostic Yield
4.3. Comprehensive Assessment of Infants with CHDs Identifies Patients in Whom a Genetic Abnormality Was Not Suspected
4.4. Incorporation of Evaluation by Medical Geneticist Increases Syndrome Diagnosis and Molecular Genetic Testing is Additive
4.5. Redundant Genetic Testing Occurs Frequently without a Clinical Algorithm
4.6. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Botto, L.D.; Lin, A.E.; Riehle-Colarusso, T.; Malik, S.; Correa, A. A National Birth Defects Prevention Study. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res. A Clin. Mol. Teratol. 2007, 79, 714–727. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic basis for congenital heart disease: Revisited. A scientific statement from the American Heart Association. Circulation 2018, 138. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.R.; Ware, S.M. Genetics and genetic testing in congenital heart disease. Clin. Perinatol. 2015, 42, 373–393. [Google Scholar] [CrossRef]
- Reuter, M.S.; Chaturvedi, R.R.; Liston, E.; Manshaei, R.; Aul, R.B.; Bowdin, S.; Cohn, I.; Curtis, M.; Dhir, P.; Hayeems, R.Z.; et al. The cardiac genome clinic: Implementing genome sequencing in pediatric heart disease. Genet. Med. 2020, 22, 1015–1024. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, T.; Tang, P.; Wu, Y.; Jiang, Z.; Dai, J.; Gu, Y.; Xu, J.; Da, M.; Ma, H.; et al. Family-based whole-genome sequencing identifies compound heterozygous protein-coding and noncoding mutations in tetralogy of Fallot. Gene 2020, 741, 144555. [Google Scholar] [CrossRef] [PubMed]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ. Res. 2019, 124, 553–563. [Google Scholar] [CrossRef]
- Alankarage, D.; Ip, E.; Szot, J.O.; Munro, J.; Blue, G.M.; Harrison, K.; Cuny, H.; Enriquez, A.; Troup, M.; Humphreys, D.T.; et al. Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genet. Med. 2018, 21, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Priest, J.R.; Osoegawa, K.; Mohammed, N.; Nanda, V.; Kundu, R.; Schultz, K.; Lammer, E.J.; Girirajan, S.; Scheetz, T.; Waggott, D.; et al. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet. 2016, 12, e1005963. [Google Scholar] [CrossRef]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef]
- Helm, B.M.; Freeze, S.L. Genetic evaluation and use of chromosome microarray in patients with isolated heart defects: Benefits and challenges of a new model in cardiovascular Care. Front. Cardiovasc. Med. 2016, 3, 19. [Google Scholar] [CrossRef]
- Lander, J.; Ware, S.M. Copy number variation in congenital heart defects. Curr. Genet. Med. Rep. 2014, 2, 168–178. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L.; et al. Genetic basis for congenital heart defects: Current knowledge: A scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef] [PubMed]
- Lammer, E.J.; Chak, J.S.; Iovannisci, D.M.; Schultz, K.; Osoegawa, K.; Yang, W.; Carmichael, S.L.; Shaw, G.M. Chromosomal abnormalities among children born with conotruncal cardiac defects. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Brueckner, M. Genetics and genomics of congenital heart disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.A.; Hinton, R.B.; Miller, E.M.; Sund, K.L.; Ruschan, J.G.; Ware, S.M. Genetic testing practices in infant with congenital heart disease. Congenit. Heart Dis. 2014, 9, 158–167. [Google Scholar] [CrossRef]
- Goldmuntz, E.; Paluru, P.; Glessner, J.; Hakonarson, H.; Biegel, J.A.; White, P.S.; Gai, X.; Shaikh, T.H. Microdeletions and microduplications in patients with congenital heart disease and multiple congenital anomalies. Congenit. Heart Dis. 2011, 6, 592–602. [Google Scholar] [CrossRef]
- Buckley, J.R.; Kavarana, M.N.; Chowdhury, S.M.; Scheurer, M.A. Current Practice and Utility of Chromosome Microarray Analysis in Infants Undergoing Cardiac Surgery. Congenit. Heart Dis. 2015, 10, E131–E138. [Google Scholar] [CrossRef]
- Wu, X.L.; Li, R.; Fu, F.; Pan, M.; Han, J.; Yang, X.; Zhang, Y.L.; Li, F.T.; Liao, C. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 2017, 17, 117. [Google Scholar] [CrossRef]
- Geddes, G.C.; Syverson, E.; Earing, M.G. Three year experience of a clinical cardiovascular genetics program for infants with congenital heart disease. Congenit. Heart Dis. 2019, 14, 832–837. [Google Scholar] [CrossRef]
- Jones, K.L.; Adam, M.P. Evaluation and diagnosis of the dysmorphic infant. Clin. Perinatol. 2015, 42, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.; Hudgins, L. The importance of minor anomalies in the evaluation of the newborn. NeoReviews 2003, 4, e99–e104. [Google Scholar] [CrossRef]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1-BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.G.; Nakev, A.; Guo, H.; Frain, S.; Tenin, G.; Liakhovitskaia, A.; Saha, P.; Priest, J.R.; Hentges, K.E.; Keavney, B.D. Association of congenital cardiovascular malformation and neuropsychiatric phenotypes with 15q11.2 (BP1-BP2) deletion in the UK Biobank. Eur. J. Hum. Genet. 2020, 28, 1265–1273. [Google Scholar] [CrossRef]
- Kuang, S.Q.; Guo, D.C.; Prakash, S.K.; McDonald, M.L.N.; Johnson, R.J.; Wang, M.; Regaldado, E.S.; Russell, L.; Cao, J.M.; Kwartler, C.; et al. GenTAC Investigators. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011, 7, e1002118. [Google Scholar] [CrossRef]
- Rigler, S.L.; Kay, D.M.; Sicko, R.J.; Fan, R.; Liu, A.; Caggana, M.; Browne, M.L.; Druschel, C.M.; Romitti, P.A.; Brody, L.C.; et al. Novel copy-number variants in a population-based investigation of classic heterotaxy. Genet. Med. 2015, 17, 348–357. [Google Scholar] [CrossRef]
- Cowan, J.R.; Tariq, M.; Shaw, C.; Rao, M.; Belmont, J.W.; Lalani, S.R.; Smolarek, T.; Ware, S.M. Copy number variation as a genetic basis for heterotaxy and heterotaxy-spectrum congential heart defects. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 371, 20150406. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, H.J.; Jacobs, K.M.; Tessier, K.M.; Narasimhan, S.L.; Tofte, A.N.; McCarter, A.R.; Cross, S.N. Chromosomal microarray analysis in the investigation of prenatally diagnosed congenital heart disease. Am. J. Obs. Gynecol. MFM 2020, 2, 100078. [Google Scholar] [CrossRef] [PubMed]
- Van Nisselrooij, A.E.L.; Lugthart, M.A.; Clur, A.-A.; Linskens, I.H.; Pajkrt, E.; Rammeloo, L.A.; Rozendaal, L.; Blom, N.A.; van Lith, J.M.M.; Knegt, A.C.; et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet. Med. 2020, 22, 1206–1214. [Google Scholar] [CrossRef]
- Hureaux, M.; Guterman, S.; Herve, B.; Till, M.; Jaillard, S.; Redon, S.; Valduga, M.; Coutton, C.; Missirian, C.; Prieur, F.; et al. Chromosomal microarray analysis in fetuses with an isolated congenital heart defect: A retrospective, nationwide, multicenter study in France. Prenat. Diag. 2019, 39, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Picker, J.; Zheng, Z.; Zhang, X.; Wang, J.; Hisama, F.; Brown, D.W.; Mullen, M.P.; Harris, D.; Stoler, J.; et al. Chromosome microarray testing for patients with congenital heart defects reveals novel disease causing loci and high diagnostic yield. BMC Genom. 2014, 15, 1127. [Google Scholar] [CrossRef]
- Thienpont, B.; Mertens, L.; de Ravel, T.; Boshoff, D.; Maas, N.; Fryns, J.P.; Gewilling, M.; Vermeesch, J.R.; Devriendt, K. Submicroscopic chromosomal imbalances detected by array-CGH area a frequent cause of congenital heart defects in selected patients. Eur. Heart J. 2007, 28, 2778–2784. [Google Scholar] [CrossRef]
- Breckpot, J.; Thienpont, B.; Peeters, H.; de Ravel, T.; Singer, A.; Rayyan, M.; Allegaert, K.; Van hole, C.; Eyskens, B.; Vermeesch, J.R.; et al. Array comparative genomic hybridization as a diagnostic tool for syndromic heart defects. J. Pediatr. 2010, 156, 810–817. [Google Scholar] [CrossRef]
- Richards, A.A.; Santos, L.J.; Nichols, H.A.; Crider, B.P.; Elder, F.F.; Hauser, N.S.; Zinn, A.R.; Garg, V. Cryptic chromosomal abnormalities identified in children with congenital heart disease. Pediatr. Res. 2008, 64, 358–363. [Google Scholar] [CrossRef]
- Erdogan, F.; Larsen, L.A.; Zhang, L.; Tumer, Z.; Tommerup, N.; Chen, W.; Jacobsen, J.R.; Schubert, M.; Jurkatis, J.; Tzschach, A.; et al. High Frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridization in patients with isolated congenital heart disease. J. Med. Genet. 2008, 45, 704–709. [Google Scholar] [CrossRef]
- Breckpot, J.; Thienpont, B.; Arens, Y.; Tranchevent, L.C.; Vermeesch, J.R.; Moreau, Y.; Gewillig, M.; Devriendt, K. Challenges in interpreting copy number variation in syndromic and non-syndromic congenital heart defects. Cytogenet. Genome Res. 2011, 135, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Ahrens-Nicklas, R.C.; Khan, S.; Garbarini, J.; Woyciechowski, S.; D’Alessandro, L.; Zackai, E.H.; Deardorff, M.A.; Goldmuntz, E. Utility of genetic evaluation in infants with congenital heart defects admitted to the cardiac intensive care unit. Am. J. Med. Genet. A 2016, 170, 3090–3097. [Google Scholar] [CrossRef] [PubMed]
- Geddes, G.C.; Basel, D.; Frommelt, P.; Kinney, A.; Earing, M. Genetic Testing Protocol Reduces Costs and Increases Rate of Genetic Diagnosis in Infants with Congenital Heart Disease. Pediatr. Cardiol. 2017, 38, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Shikany, A.R.; Landis, B.J.; Parrott, A.; Miller, E.M.; Coyan, A.; Walters, L.; Hinton, R.B.; Goldenberg, P.; Ware, S.M. A comprehensive clinical genetics approach to critical congenital heart disease in infancy. J. Pediatr. 2020, 227, 231–238. [Google Scholar] [CrossRef]
- Writing Committee for the ENIGMA-CNV Working Group; van der Meer, D.; Sønderby, I.E.; Kaufmann, T.; Walters, G.B.; Abdellaoui, A.; Ames, D.; Amunts, K.; Andersson, M.; Armstrong, N.J.; et al. Association of Copy Number Variation of the 15q11.2 BP1-BP2 Region With Cortical and Subcortical Morphology and Cognition. JAMA Psychiatry 2020, 77, 420–430. [Google Scholar] [CrossRef]
- Marchuk, D.S.; Crooks, K.; Strande, N.; Kaiser-Rogers, K.; Milko, L.; Brandt, A.; Arreola, A.; Tilley, C.R.; Bizon, C.; Vora, N.L.; et al. Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS ONE 2018, 13, e0209185. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.S.; Swint, J.M.; Lalani, S.R.; de Oliveria Otto, M.C.; Yamal, J.M.; Russell, H.V.; Lee, B.H. Exome sequencing compared with standard genetic tests for critically ill infants with suspected genetic conditions. Genet. Med. 2020, 22, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Pediatric Cardiac Genomics Consortium; Gelb, B.; Brueckner, M.; Chung, W.; Goldmuntz, E.; Kaltman, J.; Kaski, J.P.; Kim, R.; Kline, J.; Mercer-Rosa, L.; et al. The Congenital Heart Disease Genetic Network Study: Rationale, design, and early results. Circ. Res. 2013, 112, 698–706. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| CHD Class | Total Number in Study Population (%) | Completed CMA Proportion | Number (%) Syndromic (ECA-Positive) | Number (%) Apparently Isolated (ECA-Negative) |
|---|---|---|---|---|
| APVR | 14 (3.2) | 92.9% | 1 (7.1) | 13 (92.9) |
| AVSD | 13 (3.0) | 76.9% | 2 (15.4) | 11 (84.6) |
| Complex | 66 (15.0) | 83.3% | 5 (7.6) | 61 (92.4) |
| Conotruncal | 130 (29.6) | 88.5% | 46 (35.4) | 84 (64.6) |
| Heterotaxy | 35 (8.0) | 85.7% | 32 (91.4) | 3 (8.6) |
| LVOTO | 121 (27.5) | 83.5% | 24 (19.8) | 97 (80.2) |
| RVOTO | 37 (8.4) | 86.5% | 7 (18.9) | 30 (81.1) |
| Septal | 24 (5.4) | 83.3% | 15 (62.5) | 9 (37.5) |
| Total | 440 (100) | Average = 85.1% | 132 (30.0) | 308 (70.0) |
| CHD Class | Proportion of Abnormal CMA Results # | Proportion of Clinically Significant Abnormal CMA Results # |
|---|---|---|
| APVR | 23.1% (3/13) | 7.7% (1/13) |
| AVSD | 50.0% (5/10) | 20.0% (2/10) |
| Complex | 29.0% (16/55) | 10.9% (6/55) |
| Conotruncal | 28.7% (33/115) | 19.1% (22/115) |
| Heterotaxy | 20.0% (6/30) | 3.3% (2/30) * |
| LVOTO | 29.7% (30/101) | 15.8% (16/101) |
| RVOTO | 34.4% (11/32) | 9.4% (3/32) |
| Septal | 40.0% (8/20) | 20.0% (4/20) |
| Total | N = 112 | N = 55 * |
| CHD Presentation | ||||||
|---|---|---|---|---|---|---|
| Syndromic (ECA-Positive) | Apparently Isolated (ECA-Negative) | |||||
| Botto Class (Level 3) | Counts (n) | Diagnostic CMA Results (n) | Proportion Clinically Significant CMA for Class | Counts (n) | Diagnostic CMA Results (n) | Proportion Clinically Significant CMA for Class |
| APVR | 1 | 0 | 0.0% (0/1) | 13 | 1 | 7.7% (1/13) |
| AVSD | 2 | 2 | 100.0% (2/2) | 11 | 0 | 0.0% (0/11) |
| Complex | 5 | 4 | 80.0% (4/5) | 61 | 2 | 3.3% (2/61) |
| Conotruncal | 46 | 19 | 41.3% (19/46) | 84 | 3 | 3.6% (3/84) |
| Heterotaxy | 32 | 0 | 0.0% (0/32) | 3 | 1 | 33.3% (1/3) |
| LVOTO | 24 | 6 | 25.0% (6/24) | 97 | 10 | 10.3% (10/97) |
| RVOTO | 7 | 0 | 0.0% (0/7) | 30 | 3 | 10.0% (3/30) |
| Septal | 15 | 4 | 26.7% (4/15) | 9 | 0 | 0.0% (0/9) |
| Total (n) | 132 | 35 | Overall: 26.5% (35/132) | 308 | 20 | Overall: 6.5% (20/308) |
| CHD Class | Number of Clinically Significant CMAs Per CHD Class | Assessed as Low Likelihood of Genetic Abnormality | Assessed as High Likelihood of Genetic Abnormality |
|---|---|---|---|
| APVR | 1 | 1 15q11.2 deletion (BP1-BP2) | 0 |
| AVSD | 2 | 0 | 2 10 Mb duplication of 5p13.2-p11 Trisomy 21; |
| Complex | 6 | 2 22q11.2 duplication; Xp22.31 deletion | 4 2q22.1-q23.3 deletion (Mowat–Wilson syndrome, with 2% ROH and another Xq27.2 deletion); Recombinant chromosome 8 syndrome; Trisomy 18; 22q11.2 deletion |
| Conotruncal | 22 | 3 22q11.2 deletion × 2; Xq28 deletion (BRCC3, familial moyamoya); | 19 1p36 deletion syndrome; 39.31 Mb 3p22.2-pter duplication/1.68 Mb deletion of 12q24.33-qter; 6p23.2-p25.1 deletion/9q34 duplication; Trisomy 13; 16p11.2 deletion × 2; 20p12 deletion (Alagille); 22q11.2 deletion/21q22.3 duplication (5 Mb); 22q11.2 deletion/24% ROH; 22q11.2 deletion × 10 |
| Heterotaxy | 1 | 2 * 16p13.11 duplication (MYH11); 11p15.4 deletion (HBB globin cluster-carrier for β-thalassemia) * | 0 |
| LVOTO | 16 | 10 7q11.23 duplication; 8p23.1 duplication × 2; 15q11.2 deletion (BP1-BP2) × 2; 15q11.2 deletion (BP1-BP2)/1p12 duplication (NOTCH2, Alagille syndrome); 16p11.2 duplication; 17p12 deletion (PMP22, risk for neuropathy); 22q11.2 duplication; Mosaic Turner syndrome | 6 7q11.23 deletion (Williams syndrome); Trisomy 13; mosaic trisomy 13; 15q24.2-q24.3 duplication (2.2 Mb); 16p11.2 deletion × 2 |
| RVOTO | 3 | 3 16p11.2 duplication; 8p23.1 deletion; Tetrasomy X | 0 |
| Septal | 4 | 0 | 4 2q22.1-q22.3 deletion (Mowat–Wilson syndrome); Trisomy 18; 22q11.2 deletion × 2 |
| Molecular Test | Tests (N) | Proportion of Diagnostic Results | Diagnostic Findings |
|---|---|---|---|
| Heterotaxy panel | 20 | 5.0% (1/20) | DNAH11-related primary ciliary dyskinesia/heterotaxy confirmed with nasal ciliary biopsy |
| Exome | 18 | 5.6% (1/18) | IFT172-related disorder/Joubert syndrome |
| Noonan/RASopathy panel | 16 | 25% (4/16) | PTPN11 × 2, RAF1, HRAS |
| Congenital anomalies CHD panel | 26 | 0% (0/26) | N/A |
| CHD7 (CHARGE syndrome) | 4 | 5.0% (2/4) | CHD7 |
| Aortopathy panel | 2 | 0% (0/2) | N/A |
| Beckwith–Wiedemann syndrome/Russell–Silver syndrome methylation analysis | 2 | 0% (0/2) | N/A |
| ELN (nonsyndromic supravalvular aortic stenosis) | 2 | 0% (0/2) | N/A |
| Primary ciliary dyskinesia panel | 2 | 0% (0/2) | N/A |
| Cardiomyopathy panel | 1 | 0% (0/1) | N/A |
| CHD gene panel | 1 | 0% (0/1) | N/A |
| Ciliopathies panel | 1 | 0% (0/1) | N/A |
| Craniosynostosis panel | 1 | 0% (0/1) | N/A |
| Exome + mtDNA panel | 1 | 100% (1/1) | MTTL1 |
| Gonadal dysgenesis panel | 1 | 0% (0/1) | N/A |
| JAG1 (Alagille) | 1 | 100% (1/1) | JAG1 |
| Limb reduction anomalies panel | 1 | 0% (0/1) | N/A |
| SLC2A1 (Glut-1 deficiency) | 1 | 0% (0/1) | N/A |
| Total | 101 | 10 |
| Study and Dates | Size | Source | Overall Testing Yield | Chrom or FISHYield | CNV Yield | VUS | Molec Yield | CMA Testing Yield ECA vs. iCHD | Key Findings |
|---|---|---|---|---|---|---|---|---|---|
| Prenatal | |||||||||
| [28] 2011–2016 | 217 | Fetal echo database; CMA in 217/336 | 36.9% | 29.5% | 7.4% | 7.4% | N/A | ECA 64.5% iCHD 22% | Type of CHD and presence of ECA impact yield |
| [29] 2012–2016 | 919 Pre = 542 Post = 185 No testing = 192 | NL PRECOR registry; severe CHD with pre- or postnatal CMA | 30.6% | 23% | 9.9% (4.2% 22q11.2) | 2.7% CMA; 2.8% molec | 5.8% | Prenatal ECA 28.7% iCHD 11.6% | Exome seq should be offered for CHD + ECA 2nd tier if time allows |
| [30] 2015 | 239 | Cytogenetic labs; all fetuses with iCHD in France with CMA; TGA, htx, abn karyotype excluded | 7.9% (CMA) | N/A | 7.9% | 2.5% | N/A | Only iCHD | 3.1% ↑ yield even when 22q11.2 excluded; fetuses with iCHD benefit from CMA |
| Postnatal | |||||||||
| [31] 2006–2013 | 422 ECA = 260 iCHD= 162 | CMA; reasons for testing, # not tested NR; median age 7 | 21.3% | 12 cases (2.8%) found by CMA; | 15.6% for P/LP 12.8% (P only) | NR | N/A | ECA 20.6% iCHD 9.3% | CMA as 1st-tier test; among syndromic, those with DD/ID or autism ↑ yield |
| [17,32,33] | 208 | Selected syndromic CHD with CMA | Range | N/A | 6.6–20.7% | NR | N/A | ECA only | Useful testing in syndromic CHD cases without dx |
| [34] | 40 | CHD+ECA compared to iCHD, selected cohort of 20 each | 12.5% | N/A | 12.5% | NR | N/A | ECA 25% iCHD 0% | CMA identifies causes in CHD+ECA cases |
| [35,36] | 151 | CHD patients without syndromic features | Range | N/A | 3.8–4.3% | NR | N/A | iCHD only | iCHD yield less than ECA but valuable |
| [16] 2008–2010 | 277 of whom 121 had CMA | All CHD infants with cytogenetic testing (277/1087 CHD) | 15% | 14% | 3.2% cohort; 7% of CMA sub-group | 22% CMA sub-group | N/A | ECA 12% iCHD 0% | Low proportion of CHD patients tested; high rate of redundant testing. |
| [18] 2010–2013 | 275 cytogenetic testing; 535 total | All infants with critical CHD | ND | 22% (10% kayotype, 12% FISH) | 14% | 13% | N/A | NR | CMA yield highest in septal class; at least 18% redundant testing |
| [37] 2007–2012 | 364 | CICU infants with genetics consultation only (total # CHD cases NR) | 25% (9% prenatal, 16% post) | 23% (of 182 chrom); 12% (of 147 FISH/MLPA) | 9% (of 296 CMA) | 8% (of 296 CMA) | 17% (of 82 molec) | NR | CHD type influences yield; septal, AVC highest; dysmorphic features by geneticist = 7× ↑ likelihood of dx. |
| [38] Pre-protocol 2010–2014; post-protocol 2015–2016. | 733 pre 158 post | STS database infants critical CHD; post-protocol all with genetics consultation | Pre: 26% Post: 36% | Pre: 18%; FISH 9%; Post: 76% FISH 26% | Pre: 24% Post: 22% | NR | NR | NR | Multiple testing ↓ post-protocol and testing rate ↑; rate of dx ↑; cost savings |
| [39] 2010–2015 | 293 213 iCHD, 80 ECA | All infants <1 month in CICU; subset had geneticist eval | 26% | 29.1% (23/79) | 14.3% (30/210) | Included in CNV yield | 5.8% of overall cohort; 27% of those tested (17/62) | ECA 21.7%* (13/60) iCHD 11% (17/150) | CHD class, specific ECAs, dysmorphic features associated with ↑ yield. |
| [20] 2015–2018 | 201 | All infants critical CHD; excludes trisomies; all with eval by single geneticist | 33% | 17.8% (5/28) chrom 33.3% (9/27) FISH | 22.6% (43/190) | 2.1% (4/190) | 35.7% (20/56) | NR | ↑ dx rate, detection of complex phenotypes, incidental findings that alter management with inpatient cardiogenetics program; ↑ genetic testing utilization and ↓ redundant testing |
| This study 2014–2019 | 440 (376, with CMA completed) | All infants critical CHD; known chrom abn excluded; all with geneticist eval | 18% | N/A | 14.6% (55/376) | 14.9% (56/376) | 2.3% of overall cohort; 10.5% of those tested (10/95) | ECA 26.5% (35/132) iCHD 6.5% (20/308) | All CHD classes had P/LP CNVs; LVOTO often had CNVs in iCHD; conotruncal in ECA. Molecular testing additive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helm, B.M.; Landis, B.J.; Ware, S.M. Genetic Evaluation of Inpatient Neonatal and Infantile Congenital Heart Defects: New Findings and Review of the Literature. Genes 2021, 12, 1244. https://doi.org/10.3390/genes12081244
Helm BM, Landis BJ, Ware SM. Genetic Evaluation of Inpatient Neonatal and Infantile Congenital Heart Defects: New Findings and Review of the Literature. Genes. 2021; 12(8):1244. https://doi.org/10.3390/genes12081244
Chicago/Turabian StyleHelm, Benjamin M., Benjamin J. Landis, and Stephanie M. Ware. 2021. "Genetic Evaluation of Inpatient Neonatal and Infantile Congenital Heart Defects: New Findings and Review of the Literature" Genes 12, no. 8: 1244. https://doi.org/10.3390/genes12081244
APA StyleHelm, B. M., Landis, B. J., & Ware, S. M. (2021). Genetic Evaluation of Inpatient Neonatal and Infantile Congenital Heart Defects: New Findings and Review of the Literature. Genes, 12(8), 1244. https://doi.org/10.3390/genes12081244