
Genetic Diversity and Population Structure of a Rhodes Grass (Chloris gayana) Collection

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DNA Extraction and Genotyping
2.3. Data Analysis
3. Results
3.1. Informativeness and Diversity of the Markers
3.2. Genome Mapping of the DArTSeq Markers onto Reference Genomes and Selection of Genome-Wide Representative Markers
3.3. Genetic Diversity and Relationship of the Rhodes Grass Collection
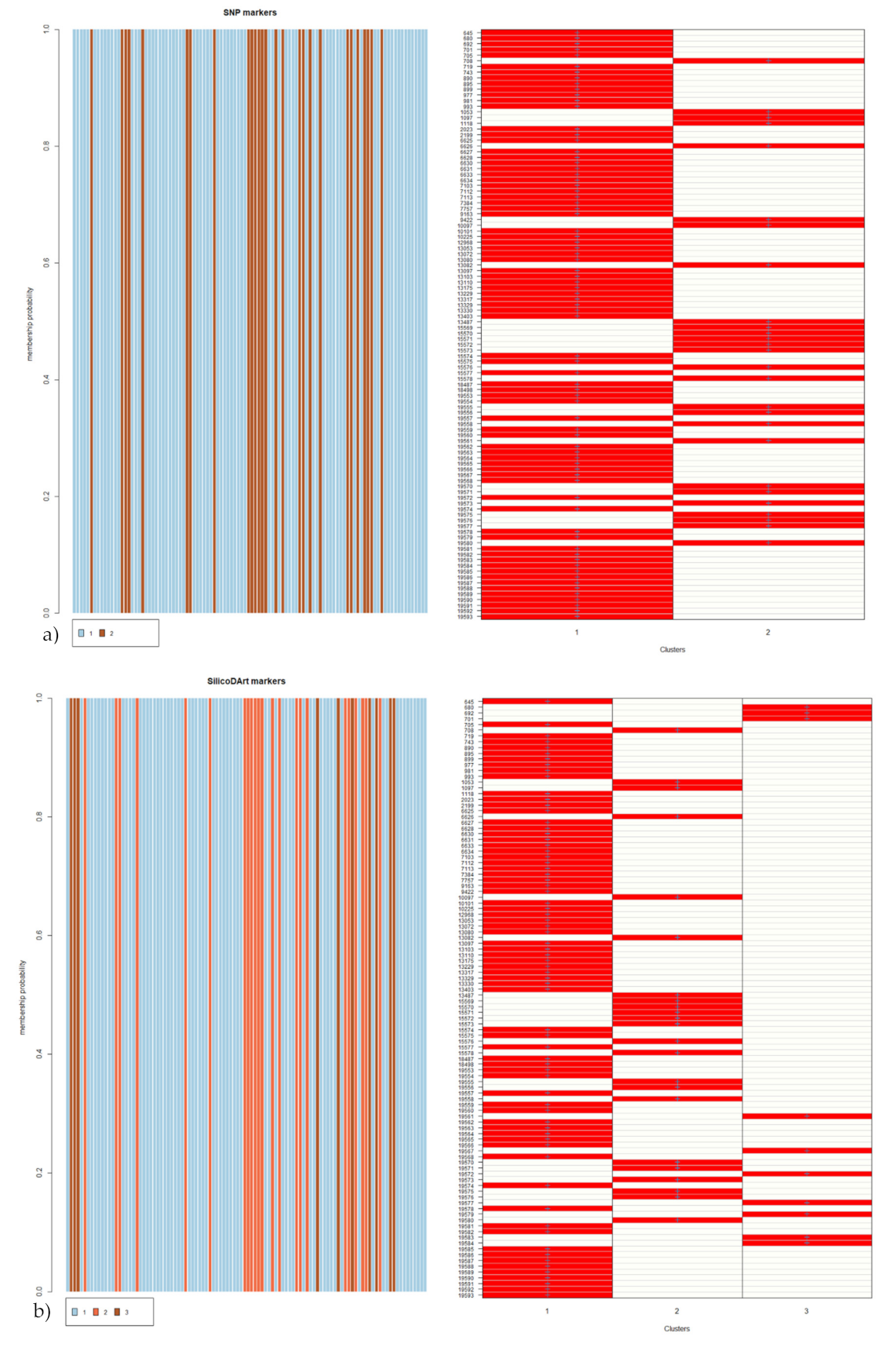
3.4. Cluster Analysis of the Collection
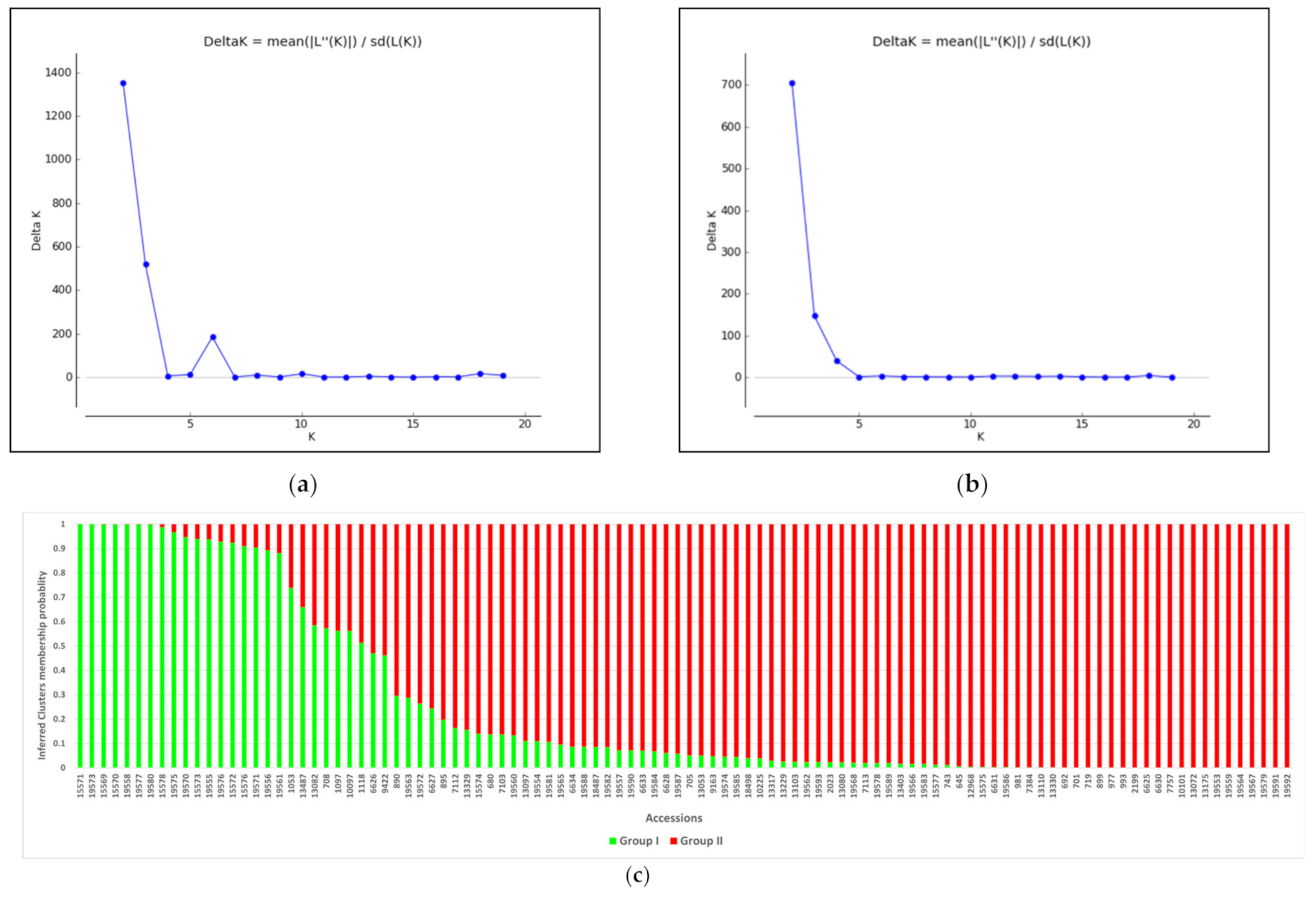
3.5. Population Structure Analysis of the Germplasm Collection
3.6. Subset Development
4. Discussion
4.1. Genotyping and Marker Diversity
4.2. Genome Wide Mapping of Markers
4.3. Genetic Diversity and Population Structure
4.4. Representative Subset Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heuzé, V.; Tran, G.; Boudon, A.; Lebas, F. Rhodes grass (Chloris gayana). Feedipedia, a programme by INRAE, CIRAD, AFZ and FAO. Last updated on 15 April 2016. Available online: https://www.feedipedia.org/node/480 (accessed on 21 April 2021).
- Cook, B.G.; Pengelly, B.C.; Schultze-Kraft, R.M.T.; Burkart, S.; Cardoso Arango, J.A.; González Guzmán, J.J.; Cox, K.; Jones, C.; Peters, M. Tropical Forages: An Interactive Selection Tool, 2nd ed.; International Center for Tropical Agriculture (CIAT), Cali, Colombia and International Livestock Research Institute (ILRI): Nairobi, Kenya, 2020; Available online: https://www.tropicalforages.info (accessed on 21 April 2020).
- Rojas-Sandoval, J. Chloris Gayana (Rhodes Grass). Invasive Species Compendium; CABI: Wallingford, UK, 2020; Available online: https://www.cabi.org/isc/datasheet/13115 (accessed on 3 May 2021).
- Liphschitz, N.; Adiva, S.I.; Eshel, A.; Waisel, Y. Salt glands on leaves of Rhodes grass (Chloris gayana Kth.). Ann. Bot. 1974, 38, 459–462. [Google Scholar] [CrossRef]
- Suttie, J.M. Hay and Straw Conservation: For Small-Scale Farming and Pastoral Conditions; Food & Agriculture Organisation: Rome, Italy, 2000. [Google Scholar]
- Masters, D.; Norman, H.; Dynes, R. Opportunities and limitations for animal production from saline land. Asian Australas. J. Anim. Sci. 2001, 14, 199–211. [Google Scholar]
- York, P.A. Range of susceptibility within and between diploid and tetraploid strains of Chloris gayana (Rhodes grass) to Meloidogyne javanica. Rev. Nématologie 1990, 13, 45–50. [Google Scholar]
- Pérez, H.; Bravo, S.; Ongaro, V.; Castagnaro, A.; Seffino, L.G.; Taleisnik, E. Chloris gayana cultivars: RAPD polymorphism and field performance under salinity. Grass Forage Sci. 1999, 54, 289–296. [Google Scholar]
- Ribotta, A.N.; López Colomba, E.; Bollati, G.P.; Striker, G.G.; Carloni, E.J.; Griffa, S.M.; Quiroga, M.P.; Tommasino, E.A.; Grunberg, K.A. Agronomic and molecular characterization of Chloris gayana cultivars and salinity response during germination and early vegetative growth. Trop. Grassl. Forrajes Trop. 2019, 7, 14–24. [Google Scholar] [CrossRef]
- Ubi, B.; Kölliker, R.; Fujimori, M.; Komatsu, T. Genetic diversity in diploid cultivars of Rhodes grass determined on the basis of amplified fragment length polymorphism markers. Crop Sci. 2003, 43, 1516–1522. [Google Scholar] [CrossRef]
- Ubi, B.E.; Fujimori, M.; Ebina, M.; Mano, Y.; Komatsu, T. AFLP variation in tetraploid cultivars of Rhodes grass (Chloris gayana Kunth). Jpn. J. Grassl. Sci. 2000, 46, 242–248. [Google Scholar]
- Ubi, B.E.; Fujimori, M.; Mano, Y.; Komatsu, T. A genetic linkage map of Rhodes grass based on an F1 pseudo-testcross population. Plant Breed. 2004, 123, 247–253. [Google Scholar] [CrossRef]
- Ribotta, A.N.; Griffa, S.M.; Díaz, D.; Carloni, E.J.; Colomba, E.L.; Tommasino, E.A.; Quiroga, M.; Luna, C.; Grunberg, K. Selecting salt-tolerant clones and evaluating genetic variability to obtain parents of new diploid and tetraploid germplasm in Rhodes grass (Chloris gayana K.). S. Afr. J. Bot. 2013, 84, 88–93. [Google Scholar] [CrossRef]
- Genesys PGR. Available online: https://www.genesys-pgr.org/ (accessed on 3 May 2021).
- Ponsens, J.; Hanson, J.; Schellberg, J.; Moeseler, B.M. Characterization of phenotypic diversity, yield and response to drought stress in a collection of Rhodes grass (Chloris gayana Kunth) accessions. Field Crop. Res. 2010, 118, 57–72. [Google Scholar] [CrossRef]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity arrays technology: A generic genome profiling technology on open platforms. Methods Mol. Biol. 2012, 888, 67–89. [Google Scholar]
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations. R Package Version 1.4.0. 2017. Available online: https://cran.r-project.org/web/packages/stringr/index.html (accessed on 12 April 2020).
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Mund, F. Factoextra: Extract and visualize the results of multivariate data analyses. 2017. Available online: https://cran.r-project.org/web/packages/factoextra/factoextra.pdf (accessed on 15 August 2019).
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; VonHoldt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- De Beukelaer, H.; Davenport, G.F.; Fack, V. Core Hunter 3: Flexible core subset selection. BMC Bioinform. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Cannarozzi, G.; Plaza-Wuthrich, S.; Esfeld, K.; Larti, S.; Wilson, Y.S.; Girma, D.; de Castro, E.; Chanyalew, S.; Blosch, R.; Farinelli, L.; et al. Genome and transcriptome sequencing identifies breeding targets in the orphan crop tef (Eragrostis tef). BMC Genom. 2014, 15, 581. [Google Scholar] [CrossRef]
- Tanaka, H.; Hirakawa, H.; Kosugi, S.; Nakayama, S.; Ono, A.; Watanabe, A.; Hashiguchi, M.; Gondo, T.; Ishigaki, G.; Muguerza, M.; et al. Sequencing and comparative analyses of the genomes of Zoysia grasses. DNA Res. 2016, 23, 171–180. [Google Scholar] [CrossRef]
- Hittalmani, S.; Mahesh, H.B.; Shirke, M.D.; Biradar, H.; Uday, G.; Aruna, Y.R.; Lohithaswa, H.C.; Mohanrao, A. Genome and transcriptome sequence of Finger millet (Eleusine coracana (L.) Gaertn.) provides insights into drought tolerance and nutraceutical properties. BMC Genom. 2017, 18, 465. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.C.; Estep, M.; Feng, L.; Vaughn, J.N.; Grimwood, J.; et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Doust, A.N.; Kellogg, E.A.; Devos, K.M.; Bennetzen, J.L. Foxtail millet: A sequence-driven grass model system. Plant Physiol. 2009, 149, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Q.; Li, J.; Zeng, C.; Hu, S.; Yu, J. The study of neighboring nucleotide composition and transition/transversion bias. Sci. China Ser. C Life Sci. 2006, 49, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.K.; Mukerji, M.; Singh, A.K.; Singh, N.K.; Mohapatra, T. SNPs in stress-responsive rice genes: Validation, genotyping, functional relevance and population structure. BMC Genom. 2012, 13, 426. [Google Scholar] [CrossRef] [PubMed]
- Feltus, F.A.; Wan, J.; Schulze, S.R.; Estill, J.C.; Jiang, N.; Paterson, A.H. An SNP resource for rice genetics and breeding based on subspecies indica and japonica genome alignments. Genome Res. 2004, 14, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Mammadov, J.; Chen, W.; Mingus, J.; Thompson, S.; Kumpatla, S. Development of versatile gene-based SNP assays in maize (Zea mays L.). Mol. Breed. 2012, 29, 779–790. [Google Scholar] [CrossRef]
- Morgil, H.; Gercek, Y.; Tulum, I. Single nucleotide polymorphisms (SNPs) in plant genetics and breeding. In The Recent Topics in Genetic Polymorphisms; Caliskan, M., Erol, O., Oz, G.C., Eds.; InTech Open: London, UK, 2020. [Google Scholar]
- Robert, F.; Pelletier, J. Exploring the mpact of single nucleotide polymorphisms on translation. Front. Genet. 2018, 9, 507. [Google Scholar] [CrossRef]
- Bohra, A.; Chand Jha, U.; Godwin, I.D.; Kumar Varshney, R. Genomic interventions for sustainable agriculture. Plant Biotechnol. J. 2020, 18, 2388–2405. [Google Scholar] [CrossRef] [PubMed]
- Thudi, M.; Palakurthi, R.; Schnable, J.C.; Chitikineni, A.; Dreisigacker, S.; Mace, E.; Srivastava, R.K.; Satyavathi, C.T.; Odeny, D.; Tiwari, V.K.; et al. Genomic resources in plant breeding for sustainable agriculture. J. Plant Physiol. 2021, 257, 153351. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Jackson, S. The first 50 plant genomes. Plant Genome 2013, 6, 2. [Google Scholar] [CrossRef]
- Negawo, A.T.; Assefa, Y.; Hanson, J.; Abdena, A.; Muktar, M.S.; Habte, E.; Sartie, A.M.; Jones, C.S. Genotyping-by-sequencing reveals population structure and genetic diversity of a Buffelgrass (Cenchrus ciliaris L.) collection. Diversity 2020, 12, 88. [Google Scholar] [CrossRef]
- Muktar, M.S.; Teshome, A.; Hanson, J.; Negawo, A.T.; Habte, E.; Domelevo Entfellner, J.-B.; Lee, K.-W.; Jones, C.S. Genotyping- by-sequencing provides new insights into the diversity of Napier grass (Cenchrus purpureus) and reveals variation in genome-wide LD patterns between collections. Sci. Rep. 2019, 9, 6936. [Google Scholar] [CrossRef] [PubMed]
- Muktar, M.S.; Habte, E.; Teshome, A.; Assefa, Y.; Negawo, A.T.; Lee, K.-W.; Zhang, J.; Jones, C.S. Insights into the genetic architecture of complex traits in Napier grass (Cenchrus purpureus) and QTL regions governing forage biomass yield, water use efficiency and feed quality traits. bioRxiv 2021. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Genomes | Number of Markers Mapped | Remark | |
|---|---|---|---|
| SilicoDArT * | SNP * | ||
| Finger millet (Eleusine coracana) | 1518 (0.8%) | 7846 (5.5%) | Scaffolds |
| Tef (Eragrostis tef) | 986 (0.5%) | 4008 (2.8%) | Scaffolds |
| Manila grass (Zoysia matrella) | 817 (0.4%) | 3554 (2.5%) | Scaffolds |
| Foxtail millet (Setaria italica) | 212 (0.1%) | 1354 (1%) | Chromosomes, scaffolds, and plastid |
| Marker Type | Source of Variation | Degree of Freedom | Sum of Square | Mean Sum of Square | Estimation Variation | Percentage of Variation | PhiPT | p Values |
|---|---|---|---|---|---|---|---|---|
| SNP | Among clusters | 1 | 4094.732 | 4094.732 | 97.418 | 33% | 0.328 | 0.000 |
| Within clusters | 102 | 20,389.393 | 199.896 | 199.896 | 67% | |||
| Total | 103 | 24,484.125 | 297.314 | 100% | ||||
| SilicoDArT | Among clusters | 2 | 4595.581 | 2297.791 | 84.988 | 30% | 0.303 | 0.000 |
| Within clusters | 101 | 19,711.217 | 195.161 | 195.161 | 70% | |||
| Total | 103 | 24,306.798 | 280.148 | 100% |
| Marker Type | Source of Variation | Degree of Freedom | Sum of Square | Mean Sum of Square | Estimation Variation | Percentage of Variation | PhiPT | p Values |
|---|---|---|---|---|---|---|---|---|
| SNP | Among subpopulation | 1 | 3888.551 | 3888.551 | 86.352 | 30% | 0.299 | 0.000 |
| Within subpopulation | 102 | 20,601.238 | 201.973 | 201.973 | 70% | |||
| Total | 103 | 24,489.788 | 288.325 | 100% | ||||
| SilicoDArT | Among subpopulation | 1 | 2309.165 | 2309.165 | 51.157 | 19% | 0.192 | 0.000 |
| Within subpopulation | 102 | 21,997.633 | 215.663 | 215.663 | 81% | |||
| Total | 103 | 24,306.798 | 266.820 | 100% |
| Accession # | Doi | Cluster | Origin |
|---|---|---|---|
| 680 | 10.18730/G6099 | I | Tanzania |
| 890 | 10.18730/G7KAE | I | Tanzania |
| 895 | 10.18730/G7KFK | I | Tanzania |
| 1118 | 10.18730/FQ2D= | II | Congo |
| 6627 | 10.18730/G5WP5 | I | South Africa |
| 6628 | 10.18730/G5WQ6 | I | Unknown |
| 6633 | 10.18730/G5WVA | I | Tanzania |
| 6634 | 10.18730/G5WWB | I | Unknown |
| 10097 | 10.18730/FP5R6 | II | Ethiopia |
| 13487 | 10.18730/FS6BT | II | Ethiopia |
| 15576 | 10.18730/FTTPU | II | Ethiopia |
| 19554 | 10.18730/FYAEQ | I | Tanzania |
| 19557 | 10.18730/FYAHT | I | Kenya |
| 19558 | 10.18730/FYAJV | II | Congo |
| 19563 | 10.18730/FYAQ * | I | Zimbabwe |
| 19568 | 10.18730/FYAW0 | II | Kenya |
| 19572 | 10.18730/FYB04 | I | India |
| 19581 | 10.18730/FYB9D | I | Tanzania |
| 19583 | 10.18730/FYBBF | I | Malawi |
| 19584 | 10.18730/FYBCG | I | Kenya |
| 19590 | 10.18730/FYBJP | I | Kenya |
| Marker Type | Source of Variation | Degree of Freedom | Sum of Square | Mean Sum of Square | Estimation Variation | Percentage of Variation | PhiPT | p Values |
|---|---|---|---|---|---|---|---|---|
| SNP | Between groups | 1 | 245.363 | 245.363 | 0.229 | 0.10% | 0.001 | 0.311 |
| Within groups | 102 | 24,244.425 | 237.690 | 237.690 | 99.90% | |||
| Total | 103 | 24,489.788 | 237.919 | 100.00% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negawo, A.T.; Muktar, M.S.; Assefa, Y.; Hanson, J.; Sartie, A.M.; Habte, E.; Jones, C.S. Genetic Diversity and Population Structure of a Rhodes Grass (Chloris gayana) Collection. Genes 2021, 12, 1233. https://doi.org/10.3390/genes12081233
Negawo AT, Muktar MS, Assefa Y, Hanson J, Sartie AM, Habte E, Jones CS. Genetic Diversity and Population Structure of a Rhodes Grass (Chloris gayana) Collection. Genes. 2021; 12(8):1233. https://doi.org/10.3390/genes12081233
Chicago/Turabian StyleNegawo, Alemayehu Teressa, Meki S. Muktar, Yilikal Assefa, Jean Hanson, Alieu M. Sartie, Ermias Habte, and Chris S. Jones. 2021. "Genetic Diversity and Population Structure of a Rhodes Grass (Chloris gayana) Collection" Genes 12, no. 8: 1233. https://doi.org/10.3390/genes12081233
APA StyleNegawo, A. T., Muktar, M. S., Assefa, Y., Hanson, J., Sartie, A. M., Habte, E., & Jones, C. S. (2021). Genetic Diversity and Population Structure of a Rhodes Grass (Chloris gayana) Collection. Genes, 12(8), 1233. https://doi.org/10.3390/genes12081233