
Guardians of the Genome: BRCA2 and Its Partners



Abstract
1. Introduction
2. BRCA2 as a Mediator of DNA DSB Repair
2.1. Role of BRCA2 in Homologous Recombination in Somatic Cells
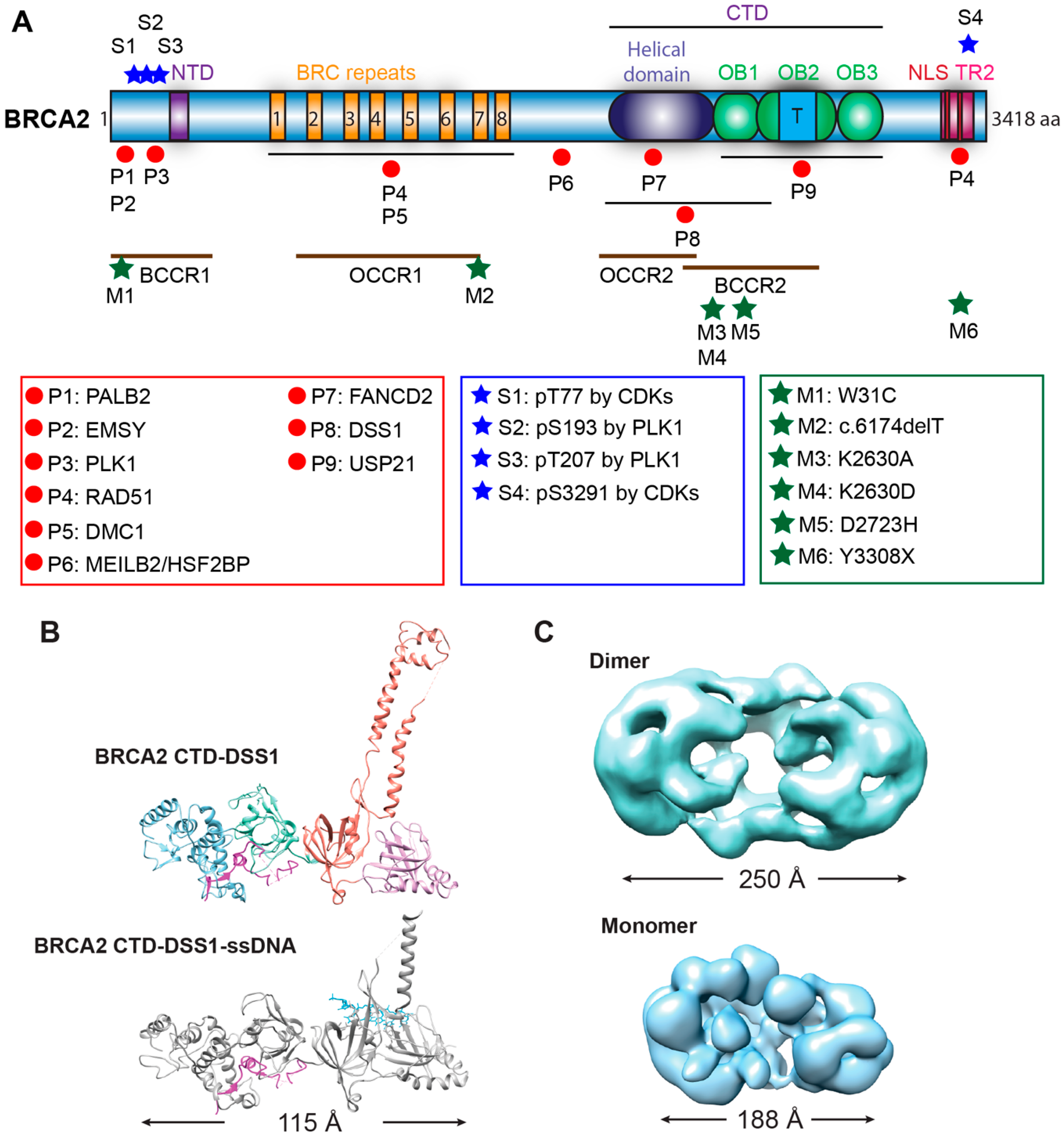
2.1.1. BRCA2 Structure
2.1.2. DNA Binding Activities of BRCA2
2.1.3. Mechanism of Nucleation of RAD51 Filaments on RPA-Coated ssDNA
2.1.4. BRCA2 Mediates RAD51 Loading
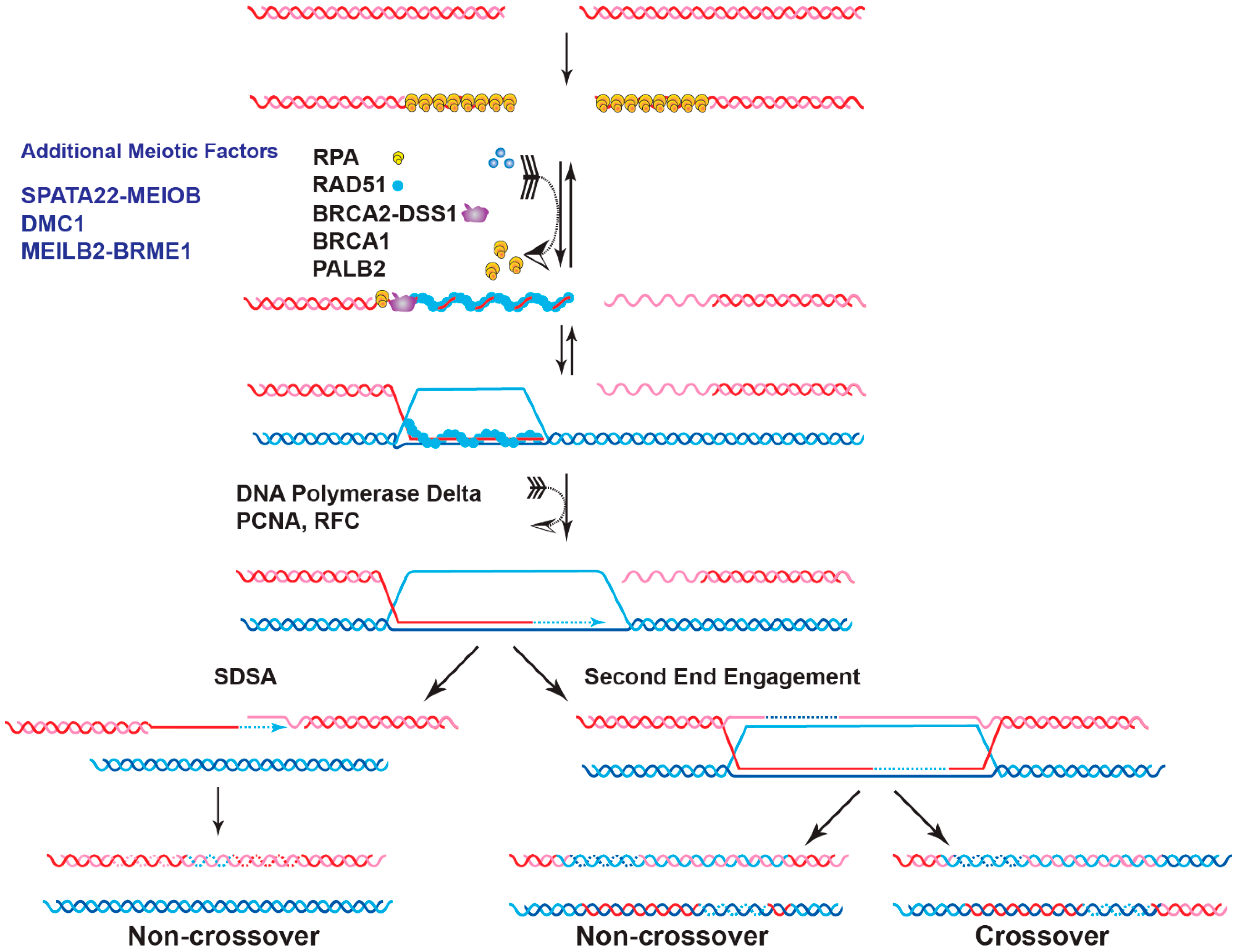
2.2. Role of BRCA2 in Meiotic Homologous Recombination
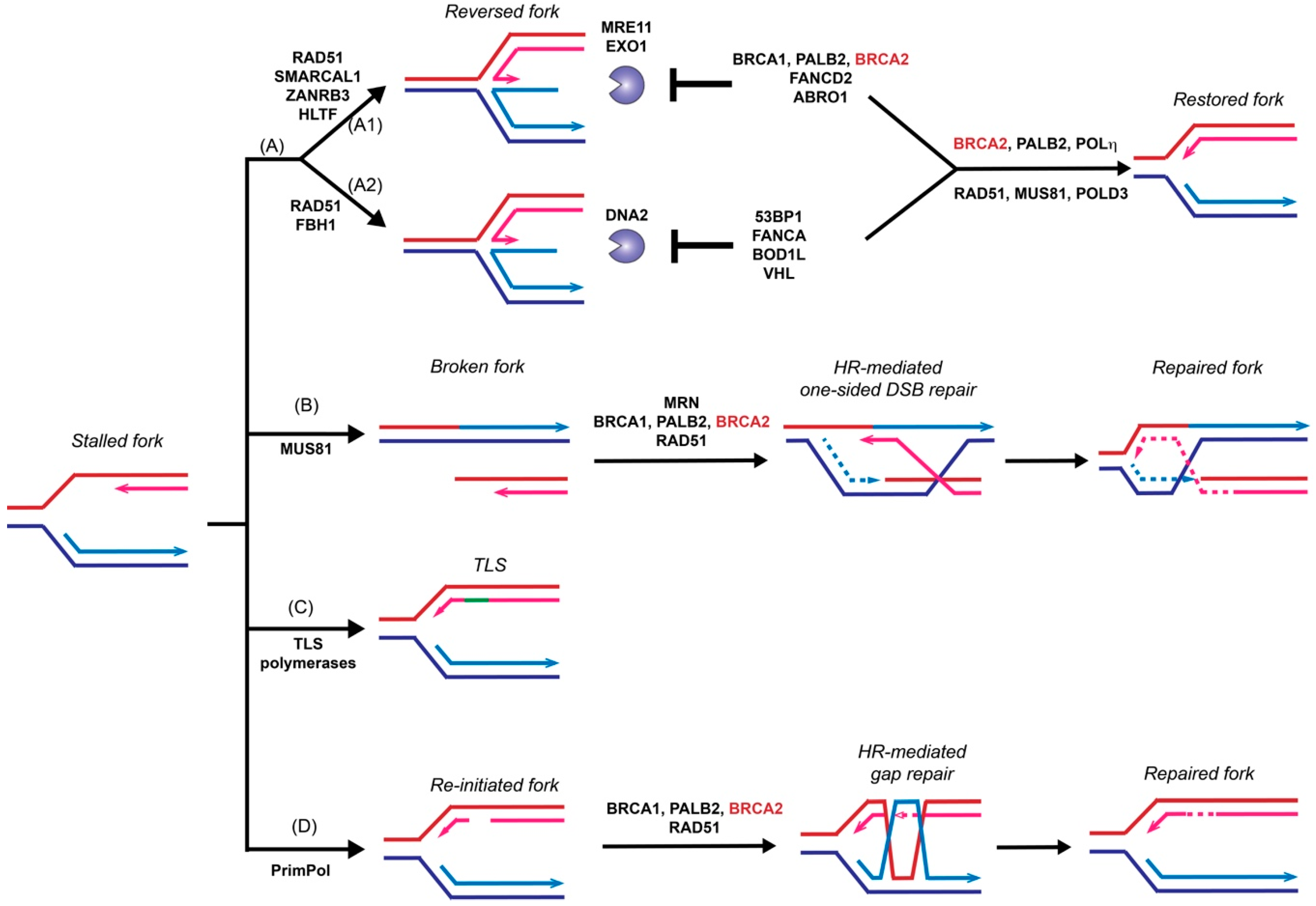
3. Role of BRCA2 in Replication Fork Restart
3.1. HR-Independent Role of BRCA2 in Replication Fork Protection
3.2. HR-Dependent Role of BRCA2 in Template Switching
4. DSB Repair Pathway Control
5. Regulation of BRCA2 Function via Its Binding Partners
5.1. DSS1, a Versatile Structural Modulator
5.2. The Partner and Localizer of BRCA2: PALB2
5.3. EMSY, a Potential Negative Regulator of BRCA2
5.4. MEILB2/HSF2BP-BRME1: A Meiotic Partner with Relevance for Cancer
5.5. SYCP3, a Cancer Testis Antigen with a Structural Role in Synaptonemal Complex
5.6. FANCD2, the Central Hub of the FA Pathway
5.7. Cyclin-Dependent Kinases
5.8. USP21
6. Pathogenic BRCA2 Mutations and BRCA2 Pseudo-Revertants
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aa | Amino acid |
| BCCR | Breast Cancer Cluster Region |
| CDK | Cyclin Dependent Kinase |
| CTD | C-terminal DNA-binding domain |
| DBD | DNA-binding domains |
| DNA | Deoxyribo Nucleic Acid |
| dsDNA | Double-stranded DNA |
| ssDNA | Single-stranded DNA |
| D-loop | Displacement loop |
| DSB | Double-Stranded Break |
| EM | Electron Microscopy |
| FA | Fanconi Anemia |
| GFP | Green Fluorescent Protein |
| HR | Homologous Recombination |
| ICL | Interstrand Crosslinks |
| IR | Irradiation |
| MMC | Mitomycin C |
| MMEJ | Microhomology-Mediated End-Joining |
| mRNA | Messenger RNA |
| NHEJ | Non-Homologous End-Joining |
| NLS | Nuclear localization signals |
| NTD | N-terminal DNA-binding domain |
| OB | Oligonucleotide/Oligosaccharide-Binding |
| OCCR | Ovarian Cancer Cluster Region |
| PARP | Poly(Adenosine diphosphate-Ribose) Polymerase |
| PARPi | PARP inhibitors |
| PIR | PARP-inhibitor-resistant |
| PLK | Polo-like Kinase |
| RFP | Replication Fork Protection |
| RNA | Ribonucleotide Acid |
| SBS | Single-Base Substitution |
| SDSA | Synthesis-Dependent Strand Annealing |
| SSA | Single-Strand Annealing |
| TLS | Translesion Synthesis |
| USP | Ubiquitin-specific Protein |
| UV | Ultraviolet |
References
- Kinzler, K.W.; Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997, 386, 761–763. [Google Scholar] [CrossRef]
- King, M.-C.; Marks, J.H.; Mandell, J.B. Breast and Ovarian Cancer Risks Due to Inherited Mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nat. Cell Biol. 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.-S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by RAD51 in mice lacking BRCA2. Nat. Cell Biol. 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.P.; Koehler, M.; Steinlein, C.; Schmid, M.; Hanakahi, L.A.; Van Gool, A.J.; West, S.; Venkitaraman, A.R. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genome Res. 2000, 14, 1400–1406. [Google Scholar]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef]
- Han, J.; Ruan, C.; Huen, M.S.Y.; Wang, J.; Xie, A.; Fu, C.; Liu, T.; Huang, J. BRCA2 antagonizes classical and alternative nonhomologous end-joining to prevent gross genomic instability. Nat. Commun. 2017, 8, 1470. [Google Scholar] [CrossRef]
- Hughes-Davies, L.; Huntsman, D.; Ruas, M.; Fuks, F.; Bye, J.; Chin, S.-F.; Milner, J.; Brown, L.; Hsu, F.; Gilks, B.; et al. EMSY Links the BRCA2 Pathway to Sporadic Breast and Ovarian Cancer. Cell 2003, 115, 523–535. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thomä, N.H.; Zheng, N.; Chen, P.-L.; Lee, W.-H.; Pavletich, N.P. BRCA2 Function in DNA Binding and Recombination from a BRCA2-DSS1-ssDNA Structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.S.; Baldeyron, C.; Carreira, A. Molding BRCA2 function through its interacting partners. Cell Cycle 2015, 14, 3389–3395. [Google Scholar] [CrossRef]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 Is Required for Homology-Directed Repair of Chromosomal Breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Xia, F.; Taghian, D.G.; DeFrank, J.S.; Zeng, Z.-C.; Willers, H.; Iliakis, G.; Powell, S.N. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc. Natl. Acad. Sci. USA 2001, 98, 8644–8649. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nat. Cell Biol. 2010, 467, 678–683. [Google Scholar] [CrossRef]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef]
- Thorslund, T.; Mcilwraith, M.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265. [Google Scholar] [CrossRef]
- Yuan, S.-S.; Lee, S.Y.; Chen, G.; Song, M.; Tomlinson, G.E.; Lee, E.Y. BRCA2 is required for ionizing radiation-induced assembly of RAD51 complex in vivo. Cancer Res. 1999, 59, 3547–3551. [Google Scholar]
- Fridlich, R.; Annamalai, D.; Roy, R.; Bernheim, G.; Powell, S.N. BRCA1 and BRCA2 protect against oxidative DNA damage converted into double-strand breaks during DNA replication. DNA Repair 2015, 30, 11–20. [Google Scholar] [CrossRef]
- Le, H.P.; Ma, X.; Vaquero, J.; Brinkmeyer, M.; Guo, F.; Heyer, W.-D.; Liu, J. DSS1 and ssDNA regulate oligomerization of BRCA2. Nucleic Acids Res. 2020, 48, 7818–7833. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Grosbart, M.; Sánchez, H.; Verhagen, B.; Van Der Zon, N.L.L.; Ristić, D.; E Van Rossum-Fikkert, S.; Wyman, C. Conformational flexibility and oligomerization of BRCA2 regions induced by RAD51 interaction. Nucleic Acids Res. 2020, 48, 9649–9659. [Google Scholar] [CrossRef]
- von Nicolai, C. A second DNA binding site in human BRCA2 promotes homologous recombination. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51–BRCA2 complex. Nat. Cell Biol. 2002, 420, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Davies, O.R.; Pellegrini, L. Interaction with the BRCA2 C terminus protects RAD51–DNA filaments from disassembly by BRC repeats. Nat. Struct. Mol. Biol. 2007, 14, 475–483. [Google Scholar] [CrossRef]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.; West, S. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef]
- Petalcorin, M.I.R.; Galkin, V.E.; Yu, X.; Egelman, E.H.; Boulton, S.J. Stabilization of RAD-51-DNA filaments via an interaction domain in Caenorhabditis elegans BRCA2. Proc. Natl. Acad. Sci. USA 2007, 104, 8299–8304. [Google Scholar] [CrossRef]
- Shahid, T.; Soroka, J.; Kong, E.H.; Malivert, L.; Mcilwraith, M.; Pape, T.; West, S.C.; Zhang, X. Structure and mechanism of action of the BRCA2 breast cancer tumor suppressor. Nat. Struct. Mol. Biol. 2014, 21, 962–968. [Google Scholar] [CrossRef]
- Reuter, M.; Zelensky, A.; Smal, I.; Meijering, E.; Van Cappellen, W.A.; De Gruiter, H.M.; van Belle, G.J.C.; van Royen, M.; Houtsmuller, A.B.; Essers, J.; et al. BRCA2 diffuses as oligomeric clusters with RAD51 and changes mobility after DNA damage in live cells. J. Cell Biol. 2014, 207, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, H.; Paul, M.W.; Grosbart, M.; Van Rossum-Fikkert, S.E.; Lebbink, J.H.G.; Kanaar, R.; Houtsmuller, A.B.; Wyman, C. Architectural plasticity of human BRCA2–RAD51 complexes in DNA break repair. Nucleic Acids Res. 2017, 45, 4507–4518. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kruswick, A.; Dang, H.; Tran, A.; Kwon, S.M.; Wang, X.W.; Oberdoerffer, P. Ubiquitin-specific protease 21 stabilizes BRCA2 to control DNA repair and tumor growth. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Oliver, A.W.; Swift, S.; Lord, C.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Ehlén, A. Proper chromosome alignment depends on BRCA2 phosphorylation by PLK1. Nat. Commun. 2020, 11, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-R.; Ting, N.S.Y.; Qin, J.; Lee, W.-H. M Phase-specific Phosphorylation of BRCA2 by Polo-like Kinase 1 Correlates with the Dissociation of the BRCA2-P/CAF Complex. J. Biol. Chem. 2003, 278, 35979–35987. [Google Scholar] [CrossRef]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC Repeats of BRCA2 Modulate the DNA-Binding Selectivity of RAD51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef]
- Carreira, A.; Kowalczykowski, S.C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 10448–10453. [Google Scholar] [CrossRef]
- Martinez, J.S. BRCA2 regulates DMC1-mediated recombination through the BRC repeats. Proc. Natl. Acad. Sci. USA 2016, 113, 3515–3520. [Google Scholar] [CrossRef]
- Zhang, J. A meiosis-specific BRCA2 binding protein recruits recombinases to DNA double-strand breaks to ensure homologous recombination. Nat. Commun. 2019, 10, 1–14. [Google Scholar]
- Brandsma, I.; Sato, K.; van Rossum-Fikkert, S.E.; van Vliet, N.; Sleddens, E.; Reuter, M.; Odijk, H.; Tempel, N.V.D.; Dekkers, D.H.; Bezstarosti, K.; et al. HSF2BP Interacts with a Conserved Domain of BRCA2 and Is Required for Mouse Spermatogenesis. Cell Rep. 2019, 27, 3790–3798.e7. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Wilson, J.B.; Medhurst, A.L.; Hejna, J.; Witt, E.; Ananth, S.; Davies, A.; Masson, J.-Y.; Moses, R.; West, S.C.; et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum. Mol. Genet. 2004, 13, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Esashi, F.; Christ, N.; Gannon, J.; Liu, Y.; Hunt, T.; Jasin, M.; West, S. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nat. Cell Biol. 2005, 434, 598–604. [Google Scholar] [CrossRef]
- Yata, K.; Bleuyard, J.-Y.; Nakato, R.; Ralf, C.; Katou, Y.; Schwab, R.A.; Niedzwiedz, W.; Shirahige, K.; Esashi, F. BRCA2 Coordinates the Activities of Cell-Cycle Kinases to Promote Genome Stability. Cell Rep. 2014, 7, 1547–1559. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of Type and Location of BRCA1 and BRCA2 Mutations with Risk of Breast and Ovarian Cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef]
- Gayther, S.A.; Mangion, J.; Russell, P.; Seal, S.; Barfoot, R.; Ponder, B.A.; Stratton, M.R.; Easton, D. Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat. Genet. 1997, 15, 103–105. [Google Scholar] [CrossRef]
- Landrum, M.J. Clin Var: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Jeyasekharan, A.; Liu, Y.; Hattori, H.; Pisupati, V.; Jonsdottir, A.B.; Rajendra, E.; Lee, M.; Sundaramoorthy, E.; Schlachter, S.; Kaminski, C.; et al. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat. Struct. Mol. Biol. 2013, 20, 1191–1198. [Google Scholar] [CrossRef]
- Siaud, N.; Barbera, M.A.; Egashira, A.; Lam, I.; Christ, N.; Schlacher, K.; Xia, B.; Jasin, M. Plasticity of BRCA2 Function in Homologous Recombination: Genetic Interactions of the PALB2 and DNA Binding Domains. PLoS Genet. 2011, 7, e1002409. [Google Scholar] [CrossRef]
- Chatterjee, G.; Jimenez-Sainz, J.; Presti, T.; Nguyen, T.; Jensen, R.B. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res. 2016, 44, 5256–5270. [Google Scholar] [CrossRef]
- Filippo, J.S.; Chi, H.-Y.; Sehorn, M.G.; Etchin, J.; Krejci, L.; Sung, P. Recombination Mediator and RAD51 Targeting Activities of a Human BRCA2 Polypeptide. J. Biol. Chem. 2006, 281, 11649–11657. [Google Scholar] [CrossRef]
- Beernink, H.T.; Morrical, S.W. RMPs: Recombination/replication mediator proteins. Trends Biochem. Sci. 1999, 24, 385–389. [Google Scholar] [CrossRef]
- Kowalczykowski, S.C. Cancer: Catalyst of a catalyst. Nature 2005, 433, 591–592. [Google Scholar] [CrossRef]
- Morimatsu, K.; Kowalczykowski, S.C. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: A universal step of recombinational repair. Mol. Cell 2003, 11, 1337–1347. [Google Scholar] [CrossRef]
- Yang, H.J. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature 2005, 433, 653–657. [Google Scholar] [CrossRef]
- Morimatsu, K.; Wu, Y.; Kowalczykowski, S.C. RecFOR Proteins Target RecA Protein to a DNA Gap with Either DNA or RNA at the 5′ Terminus Implication for Repair of Stalled Replication Forks. J. Biol. Chem. 2012, 287, 35621–35630. [Google Scholar] [CrossRef]
- Shinohara, A. Rad52 forms ring structures and co-operates with RPA in single-strand annealing. Genes Cells 1998, 3, 145–156. [Google Scholar] [CrossRef]
- Kagawa, W.; Kurumizaka, H.; Ishitani, R.; Fukai, S.; Nureki, O.; Shibata, T.; Yokoyama, S. Crystal Structure of the Homologous-Pairing Domain from the Human Rad52 Recombinase in the Undecameric Form. Mol. Cell 2002, 10, 359–371. [Google Scholar] [CrossRef]
- Singleton, M.; Wentzell, L.M.; Liu, Y.; West, S.; Wigley, D.B. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 13492–13497. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef]
- Mazin, A.V.; Zaitseva, E.; Sung, P.; Kowalczykowski, S.C. Tailed duplex DNA is the preferred substrate for RAD51 protein-mediated homologous pairing. EMBO J. 2000, 19, 1148–1156. [Google Scholar] [CrossRef]
- Baumann, P.; Benson, F.E.; West, S.C. Human RAD51 Protein Promotes ATP-Dependent Homologous Pairing and Strand Transfer Reactions In Vitro. Cell 1996, 87, 757–766. [Google Scholar] [CrossRef]
- Solinger, J.A.; Kiianitsa, K.; Heyer, W.-D. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles RAD51:dsDNA filaments. Mol. Cell 2002, 10, 1175–1188. [Google Scholar] [CrossRef]
- Sung, P.; Robberson, D.L. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell 1995, 82, 453–461. [Google Scholar] [CrossRef]
- New, J.H.; Sugiyama, T.; Zaitseva, E.; Kowalczykowski, S.C. Rad52 protein stimulates DNA strand exchange by RAD51 and replication protein A. Nat. Cell Biol. 1998, 391, 407–410. [Google Scholar] [CrossRef]
- Shinohara, A.; Ogawa, T. Stimulation by Rad52 of yeast RAD51- mediated recombination. Nat. Cell Biol. 1998, 391, 404–407. [Google Scholar] [CrossRef]
- Sung, P. Function of Yeast Rad52 Protein as a Mediator between Replication Protein A and the RAD51 Recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, A.Z. The human Rad52 protein exists as a heptameric ring. Curr. Biol. 2000, 10, 337–340. [Google Scholar] [CrossRef]
- Gibb, B.; Ye, L.F.; Kwon, Y.; Niu, H.; Sung, P.; Greene, E.C. Protein dynamics during presynaptic-complex assembly on individual single-stranded DNA molecules. Nat. Struct. Mol. Biol. 2014, 21, 893–900. [Google Scholar] [CrossRef]
- Park, M.S.; Ludwig, D.L.; Stigger, E.; Lee, S.-H. Physical Interaction between Human RAD52 and RPA Is Required for Homologous Recombination in Mammalian Cells. J. Biol. Chem. 1996, 271, 18996–19000. [Google Scholar] [CrossRef]
- Malacaria, E.; Honda, M.; Franchitto, A.; Spies, M.; Pichierri, P. Physiological and Pathological Roles of RAD52 at DNA Replication Forks. Cancers 2020, 12, 402. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kantake, N.; Wu, Y.; Kowalczykowski, S.C. Rad52-mediated DNA annealing after RAD51-mediated DNA strand exchange promotes second ssDNA capture. EMBO J. 2006, 25, 5539–5548. [Google Scholar] [CrossRef]
- Benson, F.E.; Baumann, P.; West, S. Synergistic actions of RAD51 and Rad52 in recombination and DNA repair. Nat. Cell Biol. 1998, 391, 401–404. [Google Scholar] [CrossRef]
- Liu, J.; Heyer, W.D. Who’s who in human recombination: BRCA2 and RAD52. Proc. Natl. Acad. Sci. USA 2011, 108, 441–442. [Google Scholar] [CrossRef]
- Chen, P.-L.; Chen, C.-F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.-H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292. [Google Scholar] [CrossRef]
- Wong, A.K.C.; Pero, R.; Ormonde, P.A.; Tavtigian, S.V.; Bartel, P.L. RAD51 Interacts with the Evolutionarily Conserved BRC Motifs in the Human Breast Cancer Susceptibility Gene brca2. J. Biol. Chem. 1997, 272, 31941–31944. [Google Scholar] [CrossRef]
- Stark, J.M.; Hu, P.; Pierce, A.J.; Moynahan, M.E.; Ellis, N.; Jasin, M. ATP Hydrolysis by Mammalian RAD51 Has a Key Role during Homology-directed DNA Repair. J. Biol. Chem. 2002, 277, 20185–20194. [Google Scholar] [CrossRef]
- Chen, C.-F.; Chen, P.-L.; Zhong, Q.; Sharp, Z.D.; Lee, W.-H. Expression of BRC Repeats in Breast Cancer Cells Disrupts the BRCA2-RAD51 Complex and Leads to Radiation Hypersensitivity and Loss of G2/M Checkpoint Control. J. Biol. Chem. 1999, 274, 32931–32935. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Handel, M.A.; Schimenti, J.C. Genetics of mammalian meiosis: Regulation, dynamics and impact on fertility. Nat. Rev. Genet. 2010, 11, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Hunter, N. Meiotic Recombination: The Essence of Heredity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016618. [Google Scholar] [CrossRef]
- Lam, I.; Keeney, S. Mechanism and Regulation of Meiotic Recombination Initiation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016634. [Google Scholar] [CrossRef]
- Sheridan, S.D.; Yu, X.; Roth, R.; Heuser, J.E.; Sehorn, M.G.; Sung, P.; Egelman, E.H.; Bishop, D.K. A comparative analysis of DMC1 and RAD51 nucleoprotein filaments. Nucleic Acids Res. 2008, 36, 4057–4066. [Google Scholar] [CrossRef]
- Brown, M.S.; Bishop, D.K. DNA Strand Exchange and RecA Homologs in Meiosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a016659. [Google Scholar] [CrossRef]
- Pittman, D.L.; Cobb, J.; Schimenti, K.J.; Wilson, L.A.; Cooper, D.M.; Brignull, E.; Handel, M.A.; Schimenti, J.C. Meiotic Prophase Arrest with Failure of Chromosome Synapsis in Mice Deficient for DMC1, a Germline-Specific RecA Homolog. Mol. Cell 1998, 1, 697–705. [Google Scholar] [CrossRef]
- Steinfeld, J.B.; Beláň, O.; Kwon, Y.; Terakawa, T.; Al-Zain, A.; Smith, M.J.; Crickard, J.B.; Qi, Z.; Zhao, W.; Rothstein, R.; et al. Defining the influence of RAD51 and DMC1 lineage-specific amino acids on genetic recombination. Genes Dev. 2019, 33, 1191–1207. [Google Scholar] [CrossRef]
- Lao, J.P.; Hunter, N. Trying to Avoid Your Sister. PLoS Biol. 2010, 8, e1000519. [Google Scholar] [CrossRef]
- Jones, G.H.; Franklin, F.C.H. Meiotic Crossing-over: Obligation and Interference. Cell 2006, 126, 246–248. [Google Scholar] [CrossRef]
- Connor, F.; Bertwistle, D.; Mee, P.J.; Ross, G.M.; Swift, S.; Grigorieva, E.; Tybulewicz, V.; Ashworth, A. Tumorigenesis and a DNA repair defect in mice with a truncating BRCA2 mutation. Nat. Genet. 1997, 17, 423–430. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Simard, J.; Rommens, J.M.; Couch, F.J.; Shattuck-Eidens, D.; Neuhausen, S.L.; Merajver, S.D.; Thorlacius, S.; Offit, K.; Stoppalyonnet, D.; et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat. Genet. 1996, 12, 333–337. [Google Scholar] [CrossRef]
- Chen, J.; Silver, D.P.; Walpita, D.; Cantor, S.B.; Gazdar, A.F.; Tomlinson, G.; Couch, F.J.; Weber, B.L.; Ashley, T.; Livingston, D.M.; et al. Stable Interaction between the Products of the BRCA1 and BRCA2 Tumor Suppressor Genes in Mitotic and Meiotic Cells. Mol. Cell 1998, 2, 317–328. [Google Scholar] [CrossRef]
- Sharan, S.K.; Pyle, A.; Coppola, V.; Babus, J.; Swaminathan, S.; Benedict, J.; Swing, D.; Martin, B.K.; Tessarollo, L.; Evans, J.P.; et al. BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development 2004, 131, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gurusaran, M.; Fujiwara, Y.; Zhang, K.; Echbarthi, M.; Vorontsov, E.; Guo, R.; Pendlebury, D.F.; Alam, I.; Livera, G.; et al. The BRCA2-MEILB2-BRME1 complex governs meiotic recombination and impairs the mitotic BRCA2-RAD51 function in cancer cells. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Siaud, N.; Dray, E.; Gy, I.; Gerard, E.; Takvorian, N.; Doutriaux, M.-P. BRCA2 is involved in meiosis in Arabidopsis thaliana as suggested by its interaction with DMC1. EMBO J. 2004, 23, 1392–1401. [Google Scholar] [CrossRef]
- Dray, E.; Siaud, N.; Dubois, E.; Doutriaux, M.-P. Interaction between Arabidopsis BRCA2 and Its Partners RAD51, DMC1, and DSS1. Plant Physiol. 2006, 140, 1059–1069. [Google Scholar] [CrossRef]
- Thorslund, T.; Esashi, F.; West, S.C. Interactions between human BRCA2 protein and the meiosis-specific recombinase DMC1. EMBO J. 2007, 26, 2915–2922. [Google Scholar] [CrossRef]
- Biswas, K.; Das, R.; Eggington, J.M.; Qiao, H.; North, S.L.; Stauffer, S.; Burkett, S.S.; Martin, B.K.; Southon, E.; Sizemore, S.C.; et al. Functional evaluation of BRCA2 variants mapping to the PALB2-binding and C-terminal DNA-binding domains using a mouse ES cell-based assay. Hum. Mol. Genet. 2012, 21, 3993–4006. [Google Scholar] [CrossRef]
- McAllister, K.A.; Bennett, L.M.; Houle, C.D.; Ward, T.; Malphurs, J.; Collins, N.K.; Cachafeiro, C.; Haseman, J.; Goulding, E.H.; Bunch, D.; et al. Cancer susceptibility of mice with a homozygous deletion in the COOH-terminal domain of the BRCA2 gene. Cancer Res. 2002, 62, 990–994. [Google Scholar]
- Atanassov, B.S.; Barrett, J.C.; Davis, B.J. Homozygous germ line mutation in exon 27 of murine BRCA2 disrupts the FANCD2-BRCA2 pathway in the homologous recombination-mediated DNA interstrand cross-links’ repair but does not affect meiosis. Genes Chromosom. Cancer 2005, 44, 429–437. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Zellweger, R. RAD51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Krishnamoorthy, A.; Zhao, R.; Cortez, D. Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Sci. Adv. 2020, 6, eabc3598. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Bétous, R.; Couch, F.B.; Mason, A.C.; Eichman, B.F.; Manosas, M.; Cortez, D. Substrate-Selective Repair and Restart of Replication Forks by DNA Translocases. Cell Rep. 2013, 3, 1958–1969. [Google Scholar] [CrossRef]
- Bétous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef]
- Yuan, J.; Ghosal, G.; Chen, J. The HARP-like Domain-Containing Protein AH2/ZRANB3 Binds to PCNA and Participates in Cellular Response to Replication Stress. Mol. Cell 2012, 47, 410–421. [Google Scholar] [CrossRef]
- Kile, A.C.; Chavez, D.A.; Bacal, J.; Eldirany, S.; Korzhnev, D.M.; Bezsonova, I.; Eichman, B.F.; Cimprich, K.A. HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Mol. Cell 2015, 58, 1090–1100. [Google Scholar] [CrossRef]
- Blastyák, A.; Hajdú, I.; Unk, I.; Haracska, L. Role of Double-Stranded DNA Translocase Activity of Human HLTF in Replication of Damaged DNA. Mol. Cell. Biol. 2010, 30, 684–693. [Google Scholar] [CrossRef]
- Fugger, K.; Mistrik, M.; Neelsen, K.J.; Yao, Q.; Zellweger, R.; Kousholt, A.N.; Haahr, P.; Chu, W.K.; Bartek, J.; Lopes, M.; et al. FBH1 Catalyzes Regression of Stalled Replication Forks. Cell Rep. 2015, 10, 1749–1757. [Google Scholar] [CrossRef]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Chaudhuri, A.R.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of BRCA2 and Stable RAD51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef]
- Thangavel, S. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-Dependent Degradation of Stalled DNA Replication Forks Is Prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef]
- Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022. [Google Scholar] [CrossRef]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nat. Cell Biol. 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Murphy, A.K.; Fitzgerald, M.; Ro, T.; Kim, J.H.; Rabinowitsch, A.; Chowdhury, D.; Schildkraut, C.L.; Borowiec, J.A. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J. Cell Biol. 2014, 206, 493–507. [Google Scholar] [CrossRef]
- Ayoub, N.; Rajendra, E.; Su, X.; Jeyasekharan, A.; Mahen, R.; Venkitaraman, A.R. The Carboxyl Terminus of BRCA2 Links the Disassembly of RAD51 Complexes to Mitotic Entry. Curr. Biol. 2009, 19, 1075–1085. [Google Scholar] [CrossRef]
- Morrison, C.; Shinohara, A.; Sonoda, E.; Yamaguchi-Iwai, Y.; Takata, M.; Weichselbaum, R.R.; Takeda, S. The Essential Functions of Human RAD51 Are Independent of ATP Hydrolysis. Mol. Cell. Biol. 1999, 19, 6891–6897. [Google Scholar] [CrossRef]
- Buisson, R. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase eta in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014, 6, 553–564. [Google Scholar] [CrossRef]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef]
- Higgins, N.; Kato, K.; Strauss, B. A model for replication repair in mammalian cells. J. Mol. Biol. 1976, 101, 417–425. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Davies, S.L.; Van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef]
- Adar, S.; Izhar, L.; Hendel, A.; Geacintov, N.; Livneh, Z. Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res. 2009, 37, 5737–5748. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Larminat, F.; Germanier, M.; Papouli, E.; Defais, M. Deficiency in BRCA2 leads to increase in non-conservative homologous recombination. Oncogene 2002, 21, 5188–5192. [Google Scholar] [CrossRef][Green Version]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in BRCA2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2010, 108, 686–691. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B. Signatures of mutational processes in human cancer. Nature 2018, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.; Nik-Zainal, S.; Wedge, D.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Alexandrov, L.B. The repertoire of mutational signatures in human cancer. Nat. Cell Biol. 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.; Martin, S.; Wedge, D.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nat. Cell Biol. 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Setton, J.; Reis-Filho, J.S.; Powell, S.N. Homologous recombination deficiency: How genomic signatures are generated. Curr. Opin. Genet. Dev. 2021, 66, 93–100. [Google Scholar] [CrossRef]
- Kelso, A.A.; Lopezcolorado, F.W.; Bhargava, R.; Stark, J.M. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. 2019, 15, e1008319. [Google Scholar] [CrossRef]
- Hakimi, M.-A.; Bochar, D.A.; Chenoweth, J.; Lane, W.S.; Mandel, G.; Shiekhattar, R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl. Acad. Sci. USA 2002, 99, 7420–7425. [Google Scholar] [CrossRef]
- Marmorstein, L.Y.; Kinev, A.V.; Chan, G.; Bochar, D.A.; Beniya, H.; Epstein, J.A.; Yen, T.; Shiekhattar, R. A Human BRCA2 Complex Containing a Structural DNA Binding Component Influences Cell Cycle Progression. Cell 2001, 104, 247–257. [Google Scholar] [CrossRef]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 Functionally Connects the Breast Cancer Susceptibility Proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef]
- Lee, M.; Daniels, M.; Venkitaraman, A.R. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene 2003, 23, 865–872. [Google Scholar] [CrossRef]
- Choi, E.; Park, P.-G.; Lee, H.-O.; Lee, Y.-K.; Kang, G.H.; Lee, J.W.; Han, W.; Noh, D.-Y.; Lekomtsev, S.; Lee, H. BRCA2 Fine-Tunes the Spindle Assembly Checkpoint through Reinforcement of BubR1 Acetylation. Dev. Cell 2012, 22, 295–308. [Google Scholar] [CrossRef]
- Mondal, G.; Rowley, M.; Guidugli, L.; Wu, J.; Pankratz, V.S.; Couch, F.J. BRCA2 Localization to the Midbody by Filamin a Regulates CEP55 Signaling and Completion of Cytokinesis. Dev. Cell 2012, 23, 137–152. [Google Scholar] [CrossRef]
- Takaoka, M.; Saito, H.; Takenaka, K.; Miki, Y.; Nakanishi, A. BRCA2 Phosphorylated by PLK1 Moves to the Midbody to Regulate Cytokinesis Mediated by Nonmuscle Myosin IIC. Cancer Res. 2014, 74, 1518–1528. [Google Scholar] [CrossRef]
- Cloud, V.; Chan, Y.-L.; Grubb, J.; Budke, B.; Bishop, D.K. RAD51 Is an Accessory Factor for DMC1-Mediated Joint Molecule Formation During Meiosis. Science 2012, 337, 1222–1225. [Google Scholar] [CrossRef]
- Sy, M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef]
- Ekblad, C.M. Binding of EMSY to HP1beta: Implications for recruitment of HP1beta and BS69. EMBO Rep. 2005, 6, 675–780. [Google Scholar] [CrossRef]
- Chavali, G.B.; Ekblad, C.M.; Basu, B.P.; Brissett, N.C.; Veprintsev, D.; Hughes-Davies, L.; Kouzarides, T.; Itzhaki, L.S.; Doherty, A.J. Crystal Structure of the ENT Domain of Human EMSY. J. Mol. Biol. 2005, 350, 964–973. [Google Scholar] [CrossRef]
- Vire, E.; Curtis, C.; Davalos, V.; Git, A.; Robson, S.; Villanueva, A.; Vidal, A.; Barbieri, I.; Aparicio, S.; Esteller, M.; et al. The Breast Cancer Oncogene EMSY Represses Transcription of Antimetastatic microRNA miR-31. Mol. Cell 2014, 53, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Faza, M.B.; Kemmler, S.; Jimeno, S.; González, S.J.; Aguilera, A.; Hurt, E.; Panse, V.G. Sem1 is a functional component of the nuclear pore complex–associated messenger RNA export machinery. J. Cell Biol. 2009, 184, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Ellisdon, A.; Dimitrova, L.; Hurt, E.; Stewart, M. Structural basis for the assembly and nucleic acid binding of the TREX-2 transcription-export complex. Nat. Struct. Mol. Biol. 2012, 19, 328–336. [Google Scholar] [CrossRef]
- Wilmes, G.M.; Bergkessel, M.; Bandyopadhyay, S.; Shales, M.; Braberg, H.; Cagney, G.; Collins, S.; Whitworth, G.B.; Kress, T.L.; Weissman, J.S.; et al. A Genetic Interaction Map of RNA-Processing Factors Reveals Links between Sem1/DSS1-Containing Complexes and mRNA Export and Splicing. Mol. Cell 2008, 32, 735–746. [Google Scholar] [CrossRef]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.; Tumini, E.; Moyano, E.H.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nat. Cell Biol. 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Gudmundsdottir, K.; Lord, C.; Ashworth, A. The proteasome is involved in determining differential utilization of double-strand break repair pathways. Oncogene 2007, 26, 7601–7606. [Google Scholar] [CrossRef]
- Wei, S.-J.; Williams, J.G.; Dang, H.; Darden, T.A.; Betz, B.L.; Humble, M.M.; Chang, F.-M.; Trempus, C.S.; Johnson, K.; Cannon, R.E.; et al. Identification of a Specific Motif of the DSS1 Protein Required for Proteasome Interaction and p53 Protein Degradation. J. Mol. Biol. 2008, 383, 693–712. [Google Scholar] [CrossRef] [PubMed]
- Tomko, R.J.; Hochstrasser, M. The Intrinsically Disordered Sem1 Protein Functions as a Molecular Tether during Proteasome Lid Biogenesis. Mol. Cell 2014, 53, 433–443. [Google Scholar] [CrossRef]
- Gudmundsdottir, K.; Lord, C.; Witt, E.; Tutt, A.N.J.; Ashworth, A. DSS1 is required for RAD51 focus formation and genomic stability in mammalian cells. EMBO Rep. 2004, 5, 989–993. [Google Scholar] [CrossRef]
- Kojic, M.; Yang, H.; Kostrub, C.F.; Pavletich, N.P.; Holloman, W.K. The BRCA2-Interacting Protein DSS1 Is Vital for DNA Repair, Recombination, and Genome Stability in Ustilago maydis. Mol. Cell 2003, 12, 1043–1049. [Google Scholar] [CrossRef]
- Kristensen, C.N.; Bystol, K.M.; Li, B.; Serrano, L.; Brenneman, M.A. Depletion of DSS1 protein disables homologous recombinational repair in human cells. Mutat. Res. Mol. Mech. Mutagen. 2010, 694, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zou, C.; Bai, Y.; Wazer, D.E.; Band, V.; Gao, Q. DSS1 is required for the stability of BRCA2. Oncogene 2005, 25, 1186–1194. [Google Scholar] [CrossRef]
- Marston, N.J.; Richards, W.J.; Hughes, D.; Bertwistle, D.; Marshall, C.J.; Ashworth, A. Interaction between the Product of the Breast Cancer Susceptibility Gene BRCA2 and DSS1, a Protein Functionally Conserved from Yeast to Mammals. Mol. Cell. Biol. 1999, 19, 4633–4642. [Google Scholar] [CrossRef]
- Zhao, W.; Vaithiyalingam, S.; Filippo, J.S.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol. Cell 2015, 59, 176–187. [Google Scholar] [CrossRef]
- Stefanovie, B.; Hengel, S.R.; Mlcouskova, J.; Prochazkova, J.; Spirek, M.; Nikulenkov, F.; Nemecek, D.; Koch, B.G.; Bain, F.E.; Yu, L.; et al. DSS1 interacts with and stimulates RAD52 to promote the repair of DSBs. Nucleic Acids Res. 2019, 48, 694–708. [Google Scholar] [CrossRef]
- Schenstrøm, S.M.; Rebula, C.A.; Tatham, M.H.; Hendus-Altenburger, R.; Jourdain, I.; Hay, R.T.; Kragelund, B.B.; Hartmann-Petersen, R. Expanded Interactome of the Intrinsically Disordered Protein Dss1. Cell Rep. 2018, 25, 862–870. [Google Scholar] [CrossRef]
- Sato, K.; Brandsma, I.; E Van Rossum-Fikkert, S.; Verkaik, N.; Oostra, A.B.; Dorsman, J.C.; Van Gent, D.C.; Knipscheer, P.; Kanaar, R.; Zelensky, A.N. HSF2BP negatively regulates homologous recombination in DNA interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 2442–2456. [Google Scholar] [CrossRef] [PubMed]
- Hunter, N. Synaptonemal Complexities and Commonalities. Mol. Cell 2003, 12, 533–535. [Google Scholar] [CrossRef]
- Hosoya, N.; Miyagawa, K. Synaptonemal complex proteins modulate the level of genome integrity in cancers. Cancer Sci. 2020, 112, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, N.; Okajima, M.; Kinomura, A.; Fujii, Y.; Hiyama, T.; Sun, J.; Tashiro, S.; Miyagawa, K. Synaptonemal complex protein SYCP3 impairs mitotic recombination by interfering with BRCA2. EMBO Rep. 2011, 13, 44–51. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI Protein, a Monoubiquitinated FANCD2 Paralog Required for DNA Repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, R.C.P.S.A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.B.; Yamamoto, K.; Marriott, A.S.; Hussain, S.; Sung, P.; Hoatlin, M.E.; Mathew, C.; Takata, M.; Thompson, L.H.; Kupfer, G.M.; et al. FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 and XRCC3. Oncogene 2008, 27, 3641–3652. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Andreassen, P.R.; D’Andrea, A.D. Functional Interaction of Monoubiquitinated FANCD2 and BRCA2/FANCD1 in Chromatin. Mol. Cell. Biol. 2004, 24, 5850–5862. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Michl, J.; Zimmer, J.; Buffa, F.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef]
- Kais, Z. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef]
- Tonzi, P.; Yin, Y.; Lee, C.W.T.; Rothenberg, E.; Huang, T.T. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. eLife 2018, 7, e41426. [Google Scholar] [CrossRef]
- Raghunandan, M.; Chaudhury, I.; Kelich, S.L.; Hanenberg, H.; Sobeck, A. FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi Anemia core complex. Cell Cycle 2015, 13, 342–353. [Google Scholar] [CrossRef]
- Trego, K.S.; Groesser, T.; Davalos, A.R.; Parplys, A.C.; Zhao, W.; Nelson, M.; Hlaing, A.; Shih, B.; Rydberg, B.; Pluth, J.M.; et al. Non-catalytic Roles for XPG with BRCA1 and BRCA2 in Homologous Recombination and Genome Stability. Mol. Cell 2016, 61, 535–546. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, Y.; Huan, J.; Shen, Z. Inhibition of breast and brain cancer cell growth by BCCIPα, an evolutionarily conserved nuclear protein that interacts with BRCA2. Oncogene 2001, 20, 336–345. [Google Scholar] [CrossRef]
- Lu, H.; Guo, X.; Meng, X.; Liu, J.; Allen, C.; Wray, J.; Nickoloff, J.A.; Shen, Z. The BRCA2-Interacting Protein BCCIP Functions in RAD51 and BRCA2 Focus Formation and Homologous Recombinational Repair. Mol. Cell. Biol. 2005, 25, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Kelso, A.A. The β-isoform of BCCIP promotes ADP release from the RAD51 presynaptic filament and enhances homologous DNA pairing. Nucleic Acids Res. 2017, 45, 711–725. [Google Scholar] [CrossRef]
- Huhn, S.C.; Liu, J.; Ye, C.; Lu, H.; Jiang, X.; Feng, X.; Ganesan, S.; White, E.; Shen, Z. Regulation of spindle integrity and mitotic fidelity by BCCIP. Oncogene 2017, 36, 4750–4766. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.R.; Apgar, S.; Dolios, G.; Wang, R.; Aaronson, S.A. BRCA2 Is Ubiquitinated In Vivo and Interacts with USP11, a Deubiquitinating Enzyme That Exhibits Prosurvival Function in the Cellular Response to DNA Damage. Mol. Cell. Biol. 2004, 24, 7444–7455. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Daniels, M.J.; Garnett, M.J.; Venkitaraman, A.R. A mitotic function for the high-mobility group protein HMG20b regulated by its interaction with the BRC repeats of the BRCA2 tumor suppressor. Oncogene 2011, 30, 3360–3369. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Andreeva, A.; Rutherford, T.J.; Fersht, A.R. Mapping the physical and functional interactions between the tumor suppressors p53 and BRCA2. Proc. Natl. Acad. Sci. USA 2010, 107, 8587–8592. [Google Scholar] [CrossRef]
- Zhou, Q.; Kojic, M.; Cao, Z.; Lisby, M.; Mazloum, N.A.; Holloman, W.K. DSS1 Interaction with Brh2 as a Regulatory Mechanism for Recombinational Repair. Mol. Cell. Biol. 2007, 27, 2512–2526. [Google Scholar] [CrossRef]
- Zhou, Q.; Kojic, M.; Holloman, W.K. DSS1 Release Activates DNA Binding Potential in Brh2. Biochemistry 2012, 51, 9137–9146. [Google Scholar] [CrossRef]
- Zhou, Q.; Mazloum, N.; Mao, N.; Kojic, M.; Holloman, W.K. DSS1 Regulates Interaction of Brh2 with DNA. Biochemistry 2009, 48, 11929–11938. [Google Scholar] [CrossRef]
- Zhou, Q.; Holloman, W.K. DSS1 Regulates Association of Brh2 with RAD51. Biochemistry 2017, 56, 3318–3327. [Google Scholar] [CrossRef]
- Ducy, M.; Sesma-Sanz, L.; Guitton-Sert, L.; Lashgari, A.; Gao, Y.; Brahiti, N.; Rodrigue, A.; Margaillan, G.; Caron, M.-C.; Cote, J.; et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 2019, 44, 226–240. [Google Scholar] [CrossRef]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2006, 39, 162–164. [Google Scholar] [CrossRef]
- Rahman, N.; The Breast Cancer Susceptibility Collaboration (UK); Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2006, 39, 165–167. [Google Scholar] [CrossRef]
- Xia, B.; Dorsman, J.C.; Ameziane, N.; De Vries, Y.; Rooimans, M.A.; Sheng, Q.; Pals, G.; Errami, A.; Gluckman, E.; Llera, J.; et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2006, 39, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Masson, J.-Y. PALB2 self-interaction controls homologous recombination. Nucleic Acids Res. 2012, 40, 10312–10323. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, W.; Sy, M.H.; Huen, M.S.Y. ATM-dependent Phosphorylation of the Fanconi Anemia Protein PALB2 Promotes the DNA Damage Response. J. Biol. Chem. 2015, 290, 27545–27556. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.K.; Larsen, B.; Achanta, K.; Sørensen, C.S. ATM/ATR-mediated phosphorylation of PALB 2 promotes RAD 51 function. EMBO Rep. 2016, 17, 671–681. [Google Scholar] [CrossRef]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.-L.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Noordermeer, S.; Wilson, M.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nat. Cell Biol. 2015, 528, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Luijsterburg, M.S.; Typas, D.; Caron, M.-C.; Wiegant, W.W.; Heuvel, D.V.D.; Boonen, R.A.; Couturier, A.M.; Mullenders, L.H.; Masson, J.-Y.; Van Attikum, H. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife 2017, 6, e20922. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Gil, E.R.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dray, E.; Etchin, J.; Wiese, C.; Saro, D.; Williams, G.J.; Hammel, M.; Yu, X.; Galkin, V.E.; Liu, D.; Tsai, M.-S.; et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat. Struct. Mol. Biol. 2010, 17, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Dion-Côté, A.-M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.-Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef]
- Milner, J.; Ponder, B.A.J.; Hughes-Davies, L.; Seltmann, M.; Kouzarides, T. Transcriptional activation functions in BRCA2. Nat. Cell Biol. 1997, 386, 772–773. [Google Scholar] [CrossRef]
- Cousineau, I.; Belmaaza, A. EMSY overexpression disrupts the BRCA2/RAD51 pathway in the DNA-damage response: Implications for chromosomal instability/recombination syndromes as checkpoint diseases. Mol. Genet. Genom. 2011, 285, 325–340. [Google Scholar] [CrossRef]
- Jelinic, P.; Eccles, L.A.; Tseng, J.; Cybulska, P.; Wielgos, M.; Powell, S.N.; Levine, D.A. The EMSY threonine 207 phospho-site is required for EMSY-driven suppression of DNA damage repair. Oncotarget 2017, 8, 13792–13804. [Google Scholar] [CrossRef][Green Version]
- Wilkerson, P.M.; Dedes, K.J.; Wetterskog, D.; Mackay, A.; Lambros, M.B.; Mansour, M.; Frankum, J.; Lord, C.J.; Natrajan, R.; Ashworth, A.; et al. Functional characterization of EMSY gene amplification in human cancers. J. Pathol. 2011, 225, 29–42. [Google Scholar] [CrossRef]
- Ihnen, M.; Zu Eulenburg, C.; Kolarova, T.; Qi, J.W.; Manivong, K.; Chalukya, M.; Dering, J.; Anderson, L.; Ginther, C.; Meuter, A.; et al. Therapeutic Potential of the Poly(ADP-ribose) Polymerase Inhibitor Rucaparib for the Treatment of Sporadic Human Ovarian Cancer. Mol. Cancer Ther. 2013, 12, 1002–1015. [Google Scholar] [CrossRef] [PubMed]
- Yoshima, T.; Yura, T.; Yanagi, H. Novel testis-specific protein that interacts with heat shock factor 2. Gene 1998, 214, 139–146. [Google Scholar] [CrossRef]
- Takemoto, K. Meiosis-Specific C19orf57/4930432K21Rik/BRME1 Modulates Localization of RAD51 and DMC1 to DSBs in Mouse Meiotic Recombination. Cell Rep. 2020, 31, 107686. [Google Scholar] [CrossRef]
- Felipe-Medina, N.; Caburet, S.; Sánchez-Sáez, F.; Condezo, Y.B.; de Rooij, D.G.; Gómez-H, L.; Garcia-Valiente, R.; Todeschini, A.L.; Duque, P.; Sánchez-Martin, M.A.; et al. A missense in HSF2BP causing primary ovarian insufficiency affects meiotic recombination by its novel interactor C19ORF57/BRME1. eLife 2020, 9, e56996. [Google Scholar] [CrossRef]
- Shang, Y.; Huang, T.; Liu, H.; Liu, Y.; Liang, H.; Yu, X.; Li, M.; Zhai, B.; Yang, X.; Wei, Y.; et al. MEIOK21: A new component of meiotic recombination bridges required for spermatogenesis. Nucleic Acids Res. 2020, 48, 6624–6639. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Feng, H.; Lin, Z.; Zheng, J.; Liu, D.; Guo, R.; Li, J.; Li, R.H.; Ng, E.H.; Huen, M.S.; et al. The novel male meiosis recombination regulator coordinates the progression of meiosis prophase I. J. Genet. Genom. 2020, 47, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, S.; Peterson, S.; Patel, D.; Huo, Y.; Cai, H.; Bowman-Colin, C.; Miller, S.; Ludwig, T.; Ganesan, S.; Bhaumik, M.; et al. Male Fertility Defect Associated with Disrupted BRCA1-PALB2 Interaction in Mice. J. Biol. Chem. 2014, 289, 24617–24629. [Google Scholar] [CrossRef]
- Kobayashi, W.; Hosoya, N.; Machida, S.; Miyagawa, K.; Kurumizaka, H. SYCP3 regulates strand invasion activities of RAD51 and DMC1. Genes Cells 2017, 22, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef]
- Davies, A.A.; Masson, J.-Y.; Mcilwraith, M.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in Control of the RAD51 Recombination and DNA Repair Protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Chalermrujinanant, C.; Michowski, W.; Sittithumcharee, G.; Esashi, F.; Jirawatnotai, S. Cyclin D1 promotes BRCA2-RAD51 interaction by restricting cyclin A/B-dependent BRCA2 phosphorylation. Oncogene 2015, 35, 2815–2823. [Google Scholar] [CrossRef]
- Jirawatnotai, S.; Hu, Y.; Michowski, W.; Elias, J.E.; Becks, L.; Bienvenu, F.; Zagozdzon, A.; Goswami, T.; Wang, Y.E.; Clark, A.B.; et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nat. Cell Biol. 2011, 474, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Pefani, D.-E.; Latusek, R.; Pires, I.; Grawenda, A.M.; Yee, K.S.; Hamilton, G.; Van Der Weyden, L.; Esashi, F.; Hammond, E.M.; O’Neill, E. RASSF1A–LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat. Cell Biol. 2014, 16, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Ehlén, A. The phospho-dependent role of BRCA2 on the maintenance of chromosome integrity. Cell Cycle 2021, 20, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Argunhan, B.; Iwasaki, H.; Tsubouchi, H. Post-translational modification of factors involved in homologous recombination. DNA Repair 2021, 104, 103114. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nat. Cell Biol. 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nat. Cell Biol. 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Cline, M.S. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet 2018, 14, e1007752. [Google Scholar] [CrossRef]
- Edwards, S.; Brough, R.; Lord, C.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nat. Cell Biol. 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nat. Cell Biol. 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Goggins, M.; Schutte, M.; Lu, J.; Moskaluk, C.A.; Weinstein, C.L.; Petersen, G.M.; Yeo, C.J.; Jackson, C.E.; Lynch, H.T.; Hruban, R.H.; et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996, 56, 5360–5364. [Google Scholar] [PubMed]
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011, 102, 663–669. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein | Binding Location | BRCA2-Related Function | Cellular Function | References |
|---|---|---|---|---|
| RAD51 | BRC repeats and TR2 region |
| HR RFP | [4,5,18,19,20,21,22,27,28,37,38,79,101,147] |
| DMC1 | BRC repeats and TR2 region |
| Meiotic HR | [18,39,81,83,84,85,96] |
| PALB2 | Residue 21–39 |
| HR RFP | [11,34,119,122,141,142,148] |
| EMSY | Residue 23–44 |
| HR? Transcription factor? | [10,149,150,151] |
| DSS1 | Helical + OB1 + OB2 |
| DNA repair Protein degradation RNA metabolism | [12,19,23,48,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166] |
| MEILB2 | Residues 2117–2339 |
| Meiotic HR | [40,41,93,167] |
| SYCP3 | Unknown |
| Meiotic HR | [168,169,170] |
| FANCD2 | Residues 2350–2545 |
| ICL repair RFP | [42,171,172,173,174,175,176,177,178,179] |
| FANCG | Unknown |
| ICL repair | [173] |
| XPG | Unknown |
| HR NER | [180] |
| BCCIP | Residues 2973–3001 |
| HR Chromosome segregation | [181,182,183,184] |
| Pol η | Residues 1338–1781 |
| Fork stability TLS | [122] |
| CDK2 | Residue 3291 |
| Cell-cycle control | [43,79] |
| PLK1 | Residues S193, T207 |
| Chromosome segregation Cytokinesis | [35,36,44,143,146] |
| USP21 | OB folds |
| Deubiquitylation | [33,185] |
| BRAF35 | BRC5 |
| Cytokinesis Transcriptional repression Cell-cycle progression | [139,140,186] |
| p/CAF | Residues 290–453 |
| Chromosome segregation | [144] |
| BUBR1 | Residues 3149–3418 |
| Chromosome segregation | [144] |
| Filamin A | Residues 2516–3030 |
| Cytokinesis | [145,146] |
| CEP55 | Residues 421–982 |
| Cytokinesis | [145,146] |
| Nonmuscle myosin IIC (NM-IIC) | Unknown |
| Cytokinesis | [146] |
| p53 | BRC repeats and OB2 + OB3 domains |
| Transcription control | [187] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, H.P.; Heyer, W.-D.; Liu, J. Guardians of the Genome: BRCA2 and Its Partners. Genes 2021, 12, 1229. https://doi.org/10.3390/genes12081229
Le HP, Heyer W-D, Liu J. Guardians of the Genome: BRCA2 and Its Partners. Genes. 2021; 12(8):1229. https://doi.org/10.3390/genes12081229
Chicago/Turabian StyleLe, Hang Phuong, Wolf-Dietrich Heyer, and Jie Liu. 2021. "Guardians of the Genome: BRCA2 and Its Partners" Genes 12, no. 8: 1229. https://doi.org/10.3390/genes12081229
APA StyleLe, H. P., Heyer, W.-D., & Liu, J. (2021). Guardians of the Genome: BRCA2 and Its Partners. Genes, 12(8), 1229. https://doi.org/10.3390/genes12081229