
Posterior Lissencephaly Associated with Subcortical Band Heterotopia Due to a Variation in the CEP85L Gene: A Case Report and Refining of the Phenotypic Spectrum

 , , ,
, , ,  , ,
, , {kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Whole-Exome Sequencing
2.2. mRNA Analysis
3. Results
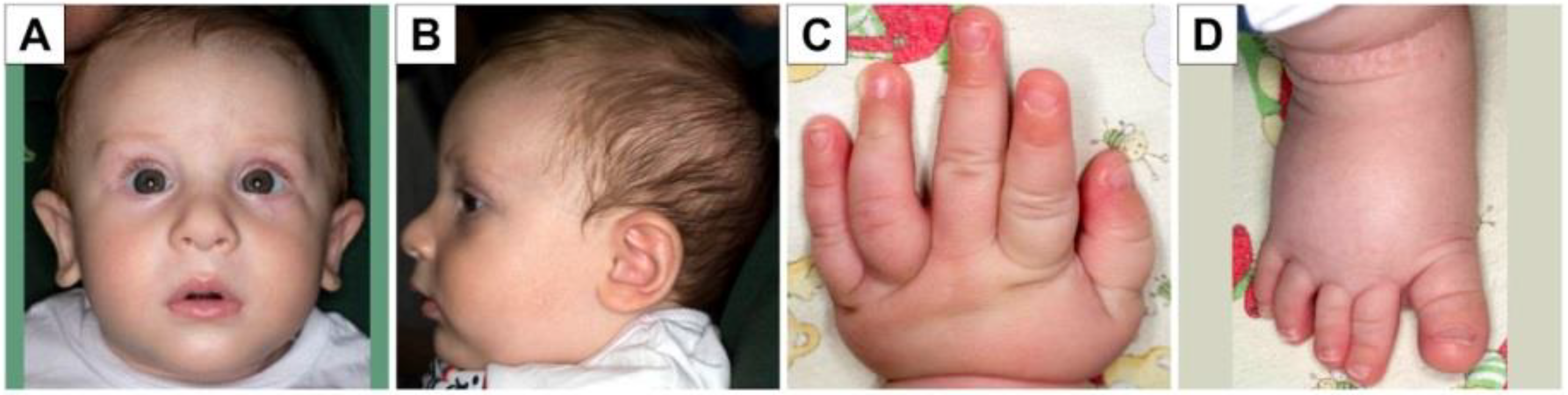
3.1. Clinical Case Presentation
3.2. Neuroradiological Imaging
3.3. EEG
3.4. Genetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guerrini, R.; Dobyns, W.B. Malformations of cortical development: Clinical features and genetic causes. Lancet Neurol. 2014, 13, 710–726. [Google Scholar] [CrossRef]
- Parrini, E.; Conti, V.; Dobyns, W.B.; Guerrini, R. Genetic basis of brain malformations. Mol. Syndr. 2016, 7, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Dobyns, W.B. Lissencephaly and the molecular basis of neuronal migration. Hum. Mol. Genet. 2003, 1, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Oegema, R.; Barakat, T.S.; Wilke, M.; Stouffs, K.; Amrom, D.; Aronica, E.; Bahi-Buisson, N.; Conti, V.; Fry, E.A.; Geis, T.; et al. International consensus recommendations on the diagnostic work-up for malformations of cortical development. Nat. Rev. Neurol. 2020, 16, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, N.; Chiari, S.; Mirzaa, G.M.; Aldinger, K.; Parrini, E.; Olds, C.; Barkovich, A.J.; Guerrini, R.; Dobyns, W.B. Lissencephaly: Expanded imaging and clinical classification. Am. J. Med. Genet. 2017, 173, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 2012, 135, 1348–1369. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.; Leventer, R.J.; Ward, H.L.; Toyo-Oka, K.; Chung, J.; Gross, A.; Ledbetter, D.H.; Martin, C.L.; Allanson, J.; Pilz, D.T.; et al. Refifinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am. J. Hum. Genet. 2003, 72, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, N.; Timms, A.E.; Aldinger, K.A.; Mirzaa, G.M.; Bennett, J.T.; Collins, S.; Olds, C.; Mei, D.; Chiari, S.; Carvill, G.; et al. Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genet. Med. 2018, 20, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Kodani, A.; Kenny, C.; Lai, A.; Gonzalez, D.M.; Stronge, E.; Sejourne, G.M.; Isacco, L.; Partlow, J.N.; O’Donnell, A.; McWalter, K.; et al. Posterior Neocortex-Specific Regulation of Neuronal Migration by CEP85L Identifies Maternal Centriole-Dependent Activation of CDK5. Neuron 2020, 106, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-H.; Muir, A.M.; Wang, W.-J.; Kang, Y.-N.; Chao, N.-H.; Wu, M.-F.; Chang, Y.-C.; Porter, B.E.; Jansen, L.A.; Sebire, G.; et al. Pathogenic variants in CEP85L cause sporadic and familial posterior predominant lissencephaly. Neuron 2020, 106, 237–245. [Google Scholar] [CrossRef] [PubMed]
- dbSNP. Available online: http://www.ncbi.nlm.nih.gov/projects/SNP (accessed on 6 January 2021).
- 1000 Genomes. A Deep Catalog of Human Genetic Variation. Available online: http://www.internationalgenome.org/ (accessed on 14 August 2020).
- Exome Variant Server. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 3 July 2020).
- ExAc. Available online: gs://gnomad-public/legacy (accessed on 3 July 2020).
- GnomAD. Available online: http://gnomad.broadinstitute.org/ (accessed on 3 July 2020).
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2018, 35, 1978. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.K.; Calligari, P.; Radio, F.C.; Caputo, V.; Dentici, M.L.; Falah, N.; High, F.-; Pantaleoni, F.; Barresi, S.; Ciolfi, A.; et al. Mutations in KCNK4 that Affect Gating Cause a Recognizable Neurodevelopmental Syndrome. Am. J. Hum. Genet. 2018, 103, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Flex, E.; Martinelli, S.; Van Dijck, A.; Ciolfi, A.; Cecchetti, S.; Coluzzi, E.; Pannone, L.; Andreoli, C.; Radio, F.C.; Pizzi, S.; et al. Aberrant Function of the C-Terminal Tail of HIST1H1E Accelerates Cellular Senescence and Causes Premature Aging. Am. J. Hum. Genet. 2019, 105, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Radio, F.C.; Pang, K.; Ciolfi, A.; Levy, M.A.; Hernández-García, A.; Pedace, L.; Pantaleoni, F.; Liu, Z.; de Boer, E.; Jackson, A.; et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 2021, 108, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Van der Auwera, G.A.; Carneiro, M.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Human Splicing Finder. Available online: http://www.umd.be/HSF/ (accessed on 24 May 2021).
- Berkeley Drosophila Genome Project—Splice Site Prediction by Neural Network. Available online: https://www.fruitfly.org/seq_tools/splice.html (accessed on 24 May 2021).
- NetGene2. Available online: http://www.cbs.dtu.dk/services/NetGene2/ (accessed on 24 May 2021).
- MaxEntScan. Available online: http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html (accessed on 24 May 2021).
- SPiCEv2.1.5. Available online: https://sourceforge.net/projects/spicev2-1/ (accessed on 6 January 2020).
- Sicca, F.; Kelemen, A.; Genton, P.; Das, S.; Mei, D.; Moro, F.; Dobyns, W.B.; Guerrini, R. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology 2003, 61, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Pagnamenta, A.T.; Lise, S.; Harrison, V.; Stewart, H.; Jayawant, S.; Quaghebeur, G.; Deng, A.T.; Murphy, V.E.; Sadighi Akha, E.; Rimmer, A.; et al. Exome sequencing can detect pathogenic mosaic mutations present at low allele frequencies. J. Hum. Genet. 2012, 57, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tian, F.; Lu, L.; Wang, Y.; Xiao, Z.; Yu, C.; Yu, X. Characterization of Cep85—A new antagonist of Nek2A that is involved in the regulation of centrosome disjunction. J. Cell Sci. 2015, 128, 3290–3303. [Google Scholar] [CrossRef] [PubMed][Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contrò, G.; Micalizzi, A.; Giangiobbe, S.; Caraffi, S.G.; Zuntini, R.; Rosato, S.; Pollazzon, M.; Terracciano, A.; Napoli, M.; Rizzi, S.; et al. Posterior Lissencephaly Associated with Subcortical Band Heterotopia Due to a Variation in the CEP85L Gene: A Case Report and Refining of the Phenotypic Spectrum. Genes 2021, 12, 1208. https://doi.org/10.3390/genes12081208
Contrò G, Micalizzi A, Giangiobbe S, Caraffi SG, Zuntini R, Rosato S, Pollazzon M, Terracciano A, Napoli M, Rizzi S, et al. Posterior Lissencephaly Associated with Subcortical Band Heterotopia Due to a Variation in the CEP85L Gene: A Case Report and Refining of the Phenotypic Spectrum. Genes. 2021; 12(8):1208. https://doi.org/10.3390/genes12081208
Chicago/Turabian StyleContrò, Gianluca, Alessia Micalizzi, Sara Giangiobbe, Stefano Giuseppe Caraffi, Roberta Zuntini, Simonetta Rosato, Marzia Pollazzon, Alessandra Terracciano, Manuela Napoli, Susanna Rizzi, and et al. 2021. "Posterior Lissencephaly Associated with Subcortical Band Heterotopia Due to a Variation in the CEP85L Gene: A Case Report and Refining of the Phenotypic Spectrum" Genes 12, no. 8: 1208. https://doi.org/10.3390/genes12081208
APA StyleContrò, G., Micalizzi, A., Giangiobbe, S., Caraffi, S. G., Zuntini, R., Rosato, S., Pollazzon, M., Terracciano, A., Napoli, M., Rizzi, S., Salerno, G. G., Radio, F. C., Niceta, M., Parrini, E., Fusco, C., Gargano, G., Guerrini, R., Tartaglia, M., Novelli, A., ... Garavelli, L. (2021). Posterior Lissencephaly Associated with Subcortical Band Heterotopia Due to a Variation in the CEP85L Gene: A Case Report and Refining of the Phenotypic Spectrum. Genes, 12(8), 1208. https://doi.org/10.3390/genes12081208