
An Integrated Clinical-Biological Approach to Identify Interindividual Variability and Atypical Phenotype-Genotype Correlations in Myopathies: Experience on A Cohort of 156 Families

 ,
,  , add
Show full author list
, add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Molecular Analyses
2.3. Bioinformatics
2.4. Variant Pathogenicity Evaluation
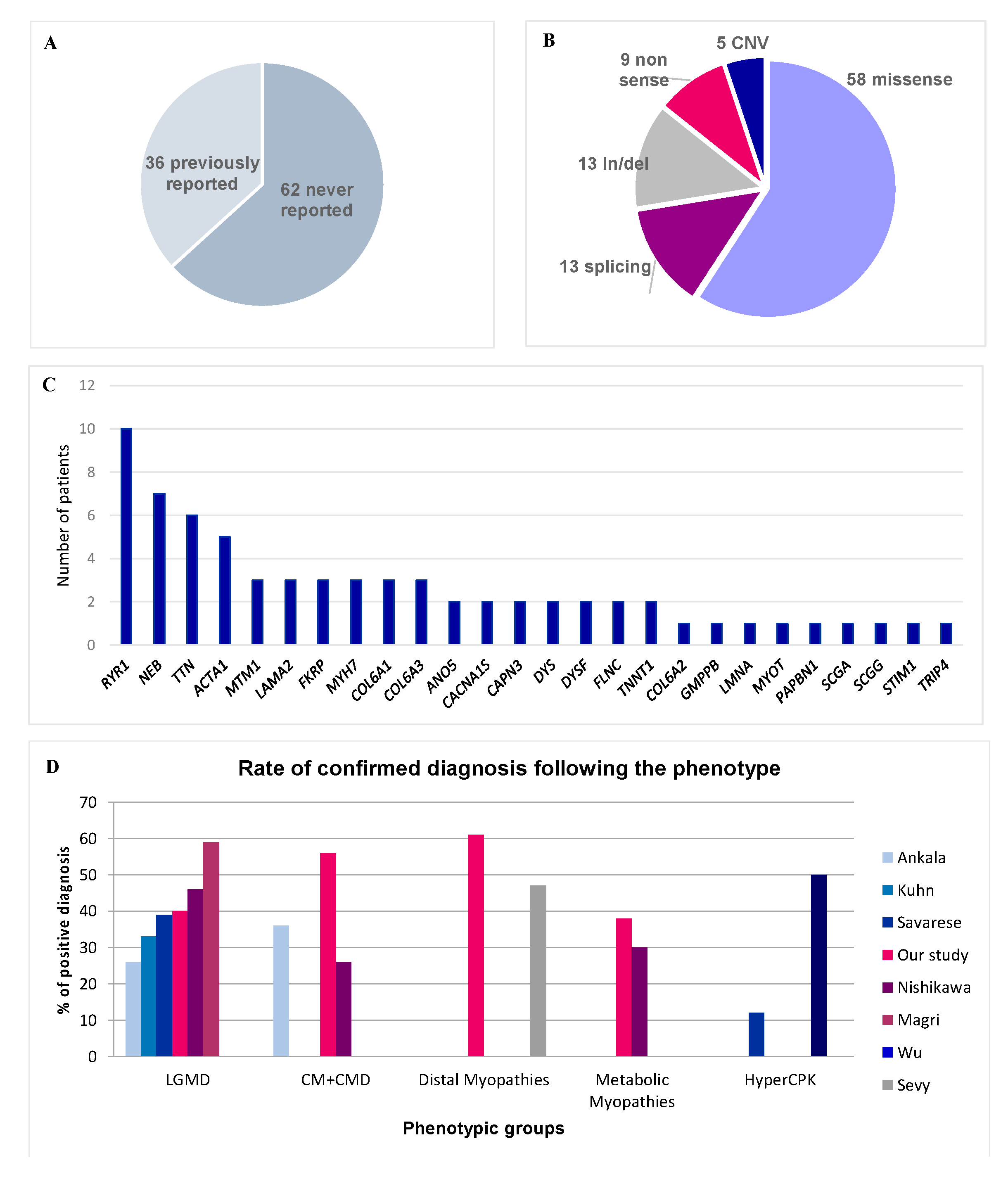
3. Results
3.1. Diagnostic Efficiency in Each Phenotypic Group
3.2. Atypical Phenotype-Genotype Associations
3.2.1. Expanded Phenotype-Genotype Associations
3.2.2. Mild Phenotypes
3.2.3. Recently Identified Phenotype-Genotype Associations
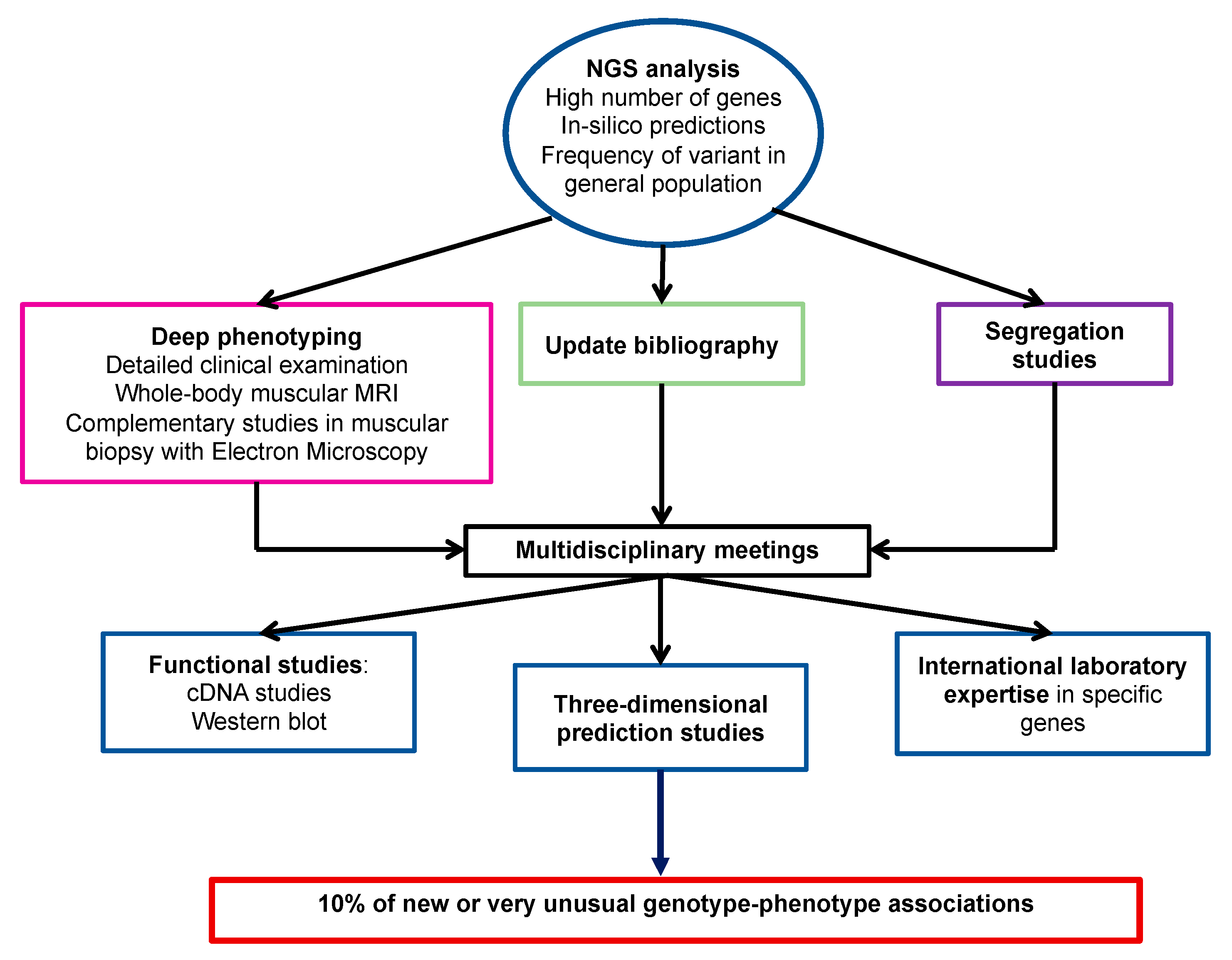
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonne, G.; Rivier, F.; Hamroun, D. The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2017, 27, 1152–1183. [Google Scholar] [CrossRef]
- Krahn, M.; Biancalana, V.; Cerino, M.; Perrin, A.; Michel-Calemard, L.; Nectoux, J.; Leturcq, F.; Bouchet-Séraphin, C.; Acquaviva-Bourdain, C.; Campana-Salort, E.; et al. A National French consensus on gene lists for the diagnosis of myopathies using next-generation sequencing. Eur. J. Hum. Genet. 2019, 27, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerve 2017, 55, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.H.; Vasta, V.; Cho, A.; Lim, B.C.; Zhang, Q.; Eun, S.H.; Hahn, S.H. Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J. Med. Genet. 2015, 52, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Brady, L.; Shoffner, J.; Tarnopolsky, M.A. Next-Generation Sequencing to Diagnose Muscular Dystrophy, Rhabdomyolysis, and HyperCKemia. Can. J. Neurol. Sci. 2018, 45, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Savarese, M.; Di Fruscio, G.; Torella, A.; Fiorillo, C.; Magri, F.; Fanin, M.; Ruggiero, L.; Ricci, G.; Astrea, G.; Passamano, L.; et al. The genetic basis of undiagnosed muscular dystrophies and myopathies. Neurology 2016, 87, 71–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, Y.; Kondo, E.; Urano, M.; Aoki, R.; Saito, K. Target resequencing of neuromuscular disease-related genes using next-generation sequencing for patients with undiagnosed early-onset neuromuscular disorders. J. Hum. Genet. 2016, 61, 931–942. [Google Scholar] [CrossRef]
- Kuhn, M.; Gläser, D.; Joshi, P.R.; Zierz, S.; Wenninger, S.; Schoser, B.; Deschauer, M. Utility of a next-generation sequencing-based gene panel investigation in German patients with genetically unclassified limb-girdle muscular dystrophy. J. Neurol. 2016, 263, 743–750. [Google Scholar] [CrossRef]
- Zenagui, R.; Lacourt, D.; Pegeot, H.; Yauy, K.; Juntas Morales, R.; Theze, C.; Rivier, F.; Cances, C.; Sole, G.; Renard, D.; et al. A Reliable Targeted Next-Generation Sequencing Strategy for Diagnosis of Myopathies and Muscular Dystrophies, Especially for the Giant Titin and Nebulin Genes. J. Mol. Diagn. 2018, 20, 533–549. [Google Scholar] [CrossRef]
- Yauy, K.; Baux, D.; Pegeot, H.; Van Goethem, C.; Mathieu, C.; Guignard, T.; Juntas Morales, R.; Lacourt, D.; Krahn, M.; Lehtokari, V.L.; et al. MoBiDiC Prioritization Algorithm, a Free, Accessible, and Efficient Pipeline for Single-Nucleotide Variant Annotation and Prioritization for Next-Generation Sequencing Routine Molecular Diagnosis. J. Mol. Diagn. 2018, 20, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Wang, J.; Yang, J.; Li, W.; Deng, Y.; Li, J.; Huang, J.; Hu, S.; Zhang, B. Comparison and evaluation of two exome capture kits and sequencing platforms for variant calling. BMC Genom. 2015, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Perrin, A.; Juntas Morales, R.; Rivier, F.; Cances, C.; Walther-Louvier, U.; Van Goethem, C.; Thèze, C.; Lacourt, D.; Pégeot, H.; Zenagui, R.; et al. The importance of an integrated genotype-phenotype strategy to unravel the molecular bases of titinopathies. Neuromuscul. Disord. 2020, 30, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Oestergaard, S.T.; Stojkovic, T.; Dahlqvist, J.R.; Bouchet-Seraphin, C.; Nectoux, J.; Leturcq, F.; Cossée, M.; Solé, G.; Thomsen, C.; Krag, T.O.; et al. Muscle involvement in limb-girdle muscular dystrophy with GMPPB deficiency (LGMD2T). Neurol. Genet. 2016, 2, e112. [Google Scholar] [CrossRef] [Green Version]
- Davignon, L.; Chauveau, C.; Julien, C.; Dill, C.; Duband-Goulet, I.; Cabet, E.; Buendia, B.; Lilienbaum, A.; Rendu, J.; Minot, M.C.; et al. The transcription coactivator ASC-1 is a regulator of skeletal myogenesis, and its deficiency causes a novel form of congenital muscle disease. Hum. Mol. Genet. 2016, 25, 1559–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knierim, E.; Hirata, H.; Wolf, N.I.; Morales-Gonzalez, S.; Schottmann, G.; Tanaka, Y.; Rudnik-Schöneborn, S.; Orgeur, M.; Zerres, K.; Vogt, S.; et al. Mutations in Subunits of the Activating Signal Cointegrator 1 Complex Are Associated with Prenatal Spinal Muscular Atrophy and Congenital Bone Fractures. Am. J. Hum. Genet. 2016, 98, 473–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar-Quiles, R.N.; Catervi, F.; Cabet, E.; Juntas-Morales, R.; Genetti, C.A.; Gidaro, T.; Koparir, A.; Yüksel, A.; Coppens, S.; Deconinck, N.; et al. ASC-1 Is a Cell Cycle Regulator Associated with Severe and Mild Forms of Myopathy. Ann. Neurol. 2020, 87, 217–232. [Google Scholar] [CrossRef]
- Richard, P.; Trollet, C.; Stojkovic, T.; De Becdelievre, A.; Perie, S.; Pouget, J.; Eymard, B. Correlation between PABPN1 genotype and disease severity in oculopharyngeal muscular dystrophy. Neurology 2017, 88, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.A.; Rubenstein, E. Proline: The Distribution, Frequency, Positioning, and Common Functional Roles of Proline and Polyproline Sequences in the Human Proteome. PLoS ONE 2013, 8, e53785. [Google Scholar] [CrossRef] [Green Version]
- Wallgren-Pettersson, C.; Lehtokari, V.L.; Kalimo, H.; Paetau, A.; Nuutinen, E.; Hackman, P.; Sewry, C.; Pelin, K.; Udd, B. Distal myopathy caused by homozygous missense mutations in the nebulin gene. Brain 2007, 130, 1465–1476. [Google Scholar] [CrossRef]
- Jain, R.K.; Jayawant, S.; Squier, W.; Muntoni, F.; Sewry, C.A.; Manzur, A.; Quinlivan, R.; Lillis, S.; Jungbluth, H.; Sparrow, J.C.; et al. Nemaline myopathy with stiffness and hypertonia associated with an acta1 mutation. Neurology 2012, 78, 1100–1103. [Google Scholar] [CrossRef]
- Von Der Ecken, J.; Müller, M.; Lehman, W.; Manstein, D.J.; Penczek, P.A.; Raunser, S. Structure of the F-actin-tropomyosin complex. Nature 2015, 519, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yu, P.; Chen, X.; Cai, T. The de novo missense mutation N117S in skeletal muscle α-actin 1 causes a mild form of congenital nemaline myopathy. Mol. Med. Rep. 2016, 14, 1693–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liewluck, T.; Sorenson, E.J.; Walkiewicz, M.A.; Rumilla, K.M.; Milone, M. Autosomal dominant distal myopathy due to a novel ACTA1 mutation. Neuromuscul. Disord. 2017, 27, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Gurel, P.S.; Kim, L.Y.; Ruijgrok, P.V.; Omabegho, T.; Bryant, Z.; Alushin, G.M. Cryo-EM structures reveal specialization at the myosin VI-actin interface and a mechanism of force sensitivity. Elife 2017, 6, e31125. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum. Mutat. 2018, 39, 1161–1172. [Google Scholar] [CrossRef]
- Schartner, V.; Romero, N.B.; Donkervoort, S.; Treves, S.; Munot, P.; Pierson, T.M.; Dabaj, I.; Malfatti, E.; Zaharieva, I.T.; Zorzato, F.; et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 2017, 133, 517–533. [Google Scholar] [CrossRef]
- Géraud, J.; Dieterich, K.; Rendu, J.; Uro Coste, E.; Dobrzynski, M.; Marcorelle, P.; Ioos, C.; Romero, N.B.; Baudou, E.; Brocard, J.; et al. Clinical phenotype and loss of the slow skeletal muscle troponin T in three new patients with recessive TNNT1 nemaline myopathy. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, W.L.; Leijenaar, J.F.; Spliet, W.G.M.; Lavrijsen, S.W.; Jansen, N.J.G.; Braun, K.P.J.; Mulder, M.; Timmers-Raaijmakers, B.; Ratsma, K.; Dooijes, D.; et al. Nemaline myopathy caused by tnnt1 mutations in a dutch pedigree. Mol. Genet. Genomic Med. 2014, 2, 134–137. [Google Scholar] [CrossRef]
- Nigro, V.; Savarese, M. Next-generation sequencing approaches for the diagnosis of skeletal muscle disorders. Curr. Opin. Neurol. 2016, 29, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.J.; Ravenscroft, G.; Laing, N.G. Skeletal muscle α-actin diseases (actinopathies): Pathology and mechanisms. Acta Neuropathol. 2013, 125, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, J.D.; Engelstad, K.E.; Ankala, A.; Tanji, K.; Dastgir, J.; De Vivo, D.C.; Coffee, B.; Chiriboga, C.A. Identification of a novel nemaline myopathy-Causing mutation in the troponin T1 (TNNT1) gene: A case outside of the old order amish. Muscle Nerve 2015, 51, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Abdulhaq, U.N.; Daana, M.; Dor, T.; Fellig, Y.; Eylon, S.; Schuelke, M.; Shaag, A.; Elpeleg, O.; Edvardson, S. Nemaline body myopathy caused by a novel mutation in troponin T1 (TNNT1). Muscle Nerve 2016, 53, 564–569. [Google Scholar] [CrossRef]
- Konersman, C.G.; Freyermuth, F.; Winder, T.L.; Lawlor, M.W.; Lagier-Tourenne, C.; Patel, S.B. Novel autosomal dominant TNNT1 mutation causing nemaline myopathy. Mol. Genet. Genom. Med. 2017, 5, 678–691. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Sex, Age/ Age at Onset | Clinical Features | CK | Muscle Biopsy | Muscle MRI | Family History | Genetic Testing |
|---|---|---|---|---|---|---|
| I298 M, 38y/ neonatal | Neonatal hypotonia Scoliosis Progressive myopathy with contractures Early-onset cardiomyopathy Restrictive respiratory syndrome | N | Type I fiber predominance. Cytoplasmic bodies | ND | Sporadic | c.3557C>T; p.(Ala1186Val) FLNC NM_001458.4 Missense de novo variant Absent in Exac and gnomAD |
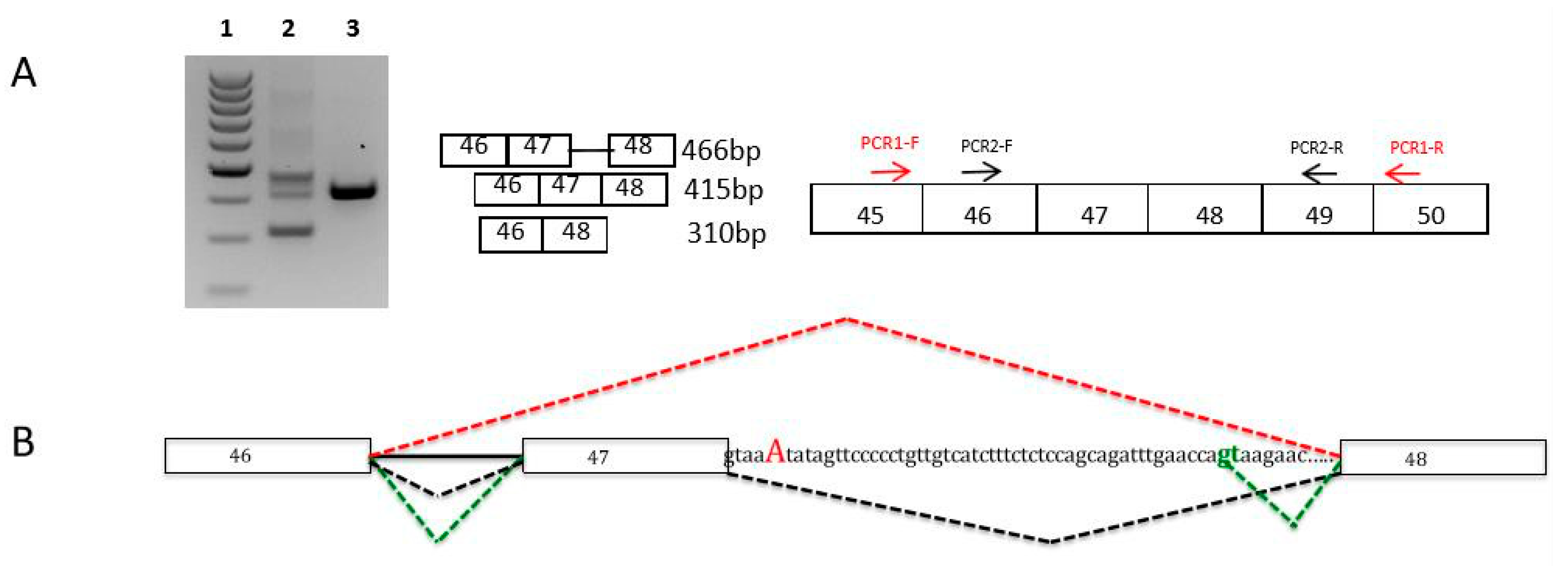
| I111 F, 4y/ 1y | Pierre Robin sequence (micrognathia, glossoptosis, and cleft palate), scoliosis Feeding difficulties Severe restrictive cardiomyopathy Mild proximal and axial weakness | N | Type I fiber atrophy | Normal | Sporadic | c.4927+2T>A FLNC de novo splicing variant HSF, MaxEnt: -100% Absent in Exac and gnomAD |
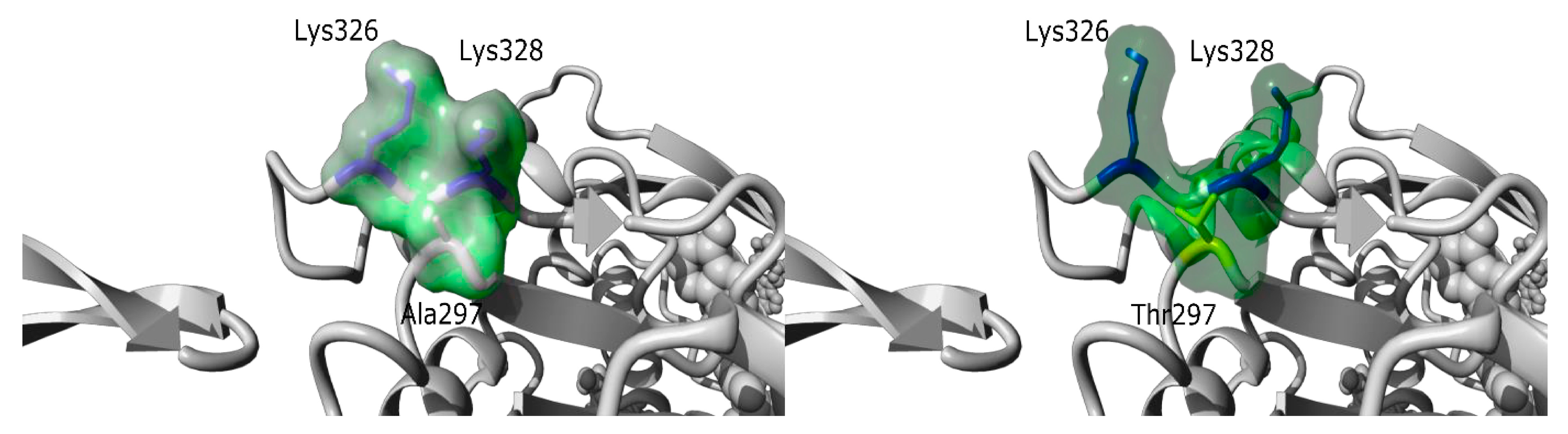
| I192 M, 20y/ 20y | Asymptomatic CK elevation Strictly normal neurological examination | x20 | Some necrotic fibers Absence of rods | Normal | Sporadic | c.889G>A; p.(Ala297Thr) ACTA1 NM_001100.3 de novo missense variant. Absent in Exac and gnomAD |
| I14 F, 75y/ 60y | Slowly progressive axial weakness with camptocormia Mild lower limb proximal weakness No ptosis, no dysphagia | N | Abnormal myofibrillar network Rimmed and not rimmed vacuoles | Paravertebral VL, AB, AM, SM, GM, SO | Sporadic | c.30_31insGCA; p.(Ala11dup) PAPBN1 NM_004643.3 |
| I303 F, 30y/ neonatal | Neonatal hypotonia with feeding difficulties and respiratory insufficiency Progressive improvement Last follow-up visit: mild axial and proximal weakness Gowers + Distal hyperlaxity Worsening with fever | N | Non-specific myopathic pattern with type 1 fiber predominance and mild myofibrillar disorganization | Upper and lower limbs atrophy. No fat tissue replacement | AR (deceased affected brother) | c.2970G>A; p.(Trp990*) CACNA1S c.5104C>T; p.(Arg1702*) CACNA1S NM_000069.2 |
| I164 M, 21y/ neonatal | Neonatal hypotonia, feeding difficulties, respiratory insufficiency Last follow-up visit: tongue deviation, mild bilateral facial paresis, mild bilateral scapula alata, proximal lower limb weakness, distal hyperlaxity | N | Non-specific myopathic pattern with type 1 fiber predominance | Tongue GMax | Pauci symptomatic father carries the variant | c.2447T>G; p.(Leu816Arg) CACNA1S Missense variant predicted to be deleterious Absent in Exac and gnomAD |
| I142 F, 40y/ 35y | Bilateral calf and left tibialis anterior atrophy | N | Excessive internal nuclei. Minicores with NADH technique | Bilateral GM and left TA | AD | del <11-18> exons TTN NM_001267550.1 |
| I26 M, 14y/ 3y | Very mild proximal weakness Mild facial paresis Moderate restrictive respiratory syndrome | N | Excessive internal nuclei resembling centronuclear myopathy. Minicores revealed by NADH staining | Normal | Adopted child | c.51437-4_51444del TTN c.26503A>T; p.(Lys8835*) TTN |
| I47 F, 44y/ 4y | Proximal and axial weakness Wheelchair at the age of 17 Last follow-up visit: severe proximal and axial weakness, restrictive respiratory syndrome | N | Excessive internal nuclei resembling centronuclear myopathy. Minicores revealed by NADH staining | Bilateral SO and left TA | Sporadic | c.65575+2T>G TTN c.68029A>G; p.(Thr22677Ala) TTN Missense variant predicted to affect splicing |
| I74, I73 F, 4y/ 3y | Mild proximal and axial weakness Facial paresis | x2 | Excessive internal nuclei Minicores in NADH staining. Abnormal western blot results: absence of the C-terminal part of TTN (anti-TTN M10-1 antibody); absence of calpain | N.D | Affected twin sister | c.106531G>C; p.(Ala35511Pro) TTN Missense variant predicted to affect splicing c.105036C>A; p.(Tyr35012*) TTN |
| I76, I77 M, 54y F, 50y/ 35y | Isolated tibialis anterior weakness Recently the brother developed finger extensor and neck flexor weakness | N | Pathological fiber size variation. Excessive internal nuclei Absence of rods | Bilateral TA and GM in both patients | AR | c.8860delG p.(Ala2954Profs*8) NEB NM_001271208.1 c.21928T>C; p.(Ser7310Pro) NEB |
| I112 M, 60y/ 57y | Isolated tibialis anterior weakness | N | Pathological fiber size variation. Absence of rods | Bilateral TA and left SO | Sporadic | c.21790G>C p.(Arg7264His) NEB c.194C>T p.(Pro65Leu) NEB |
| I60 M, 61y/ 20y | Axial and proximal weakness Retrognathia, pectus excavatum Mild rigid spine Dilated cardiomyopathy | N | Excessive internal nuclei Minicore-like lesions, rods | Bilateral SM, BF, SO | Sporadic (Consang) | del <8-9> homozygous exons TRIP4 NM_016213.4 In-Frame deletion of exons 8 and 9 |
| I172 F, 11y/ neonatal | Neonatal hypotonia Proximal and axial weakness in childhood. Gowers + Mild tibialis anterior weakness Scapula alata Facial paresis | N | Excessive internal nuclei | Bilateral GMax, AM, SM, BF, TA, SO, P | Sporadic | c.200A>G; p.(His67Arg) TNNT1 NM_003283.5 de novo missense variant Absent in Exac and gnomAD |
| I152 M, 35y/ 20y | Lower limb-girdle muscular dystrophy | x20 | Non-specific dystrophic pattern | Bilateral VL, AM, AB, SM, ST, GM | AR Affected sister. Consang | c.6617C>T; p.(Thr2206Met) Homozygous RYR1 NM_000540.2 Missense variant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juntas Morales, R.; Perrin, A.; Solé, G.; Lacourt, D.; Pegeot, H.; Walther-Louvier, U.; Cintas, P.; Cances, C.; Espil, C.; Theze, C.; et al. An Integrated Clinical-Biological Approach to Identify Interindividual Variability and Atypical Phenotype-Genotype Correlations in Myopathies: Experience on A Cohort of 156 Families. Genes 2021, 12, 1199. https://doi.org/10.3390/genes12081199
Juntas Morales R, Perrin A, Solé G, Lacourt D, Pegeot H, Walther-Louvier U, Cintas P, Cances C, Espil C, Theze C, et al. An Integrated Clinical-Biological Approach to Identify Interindividual Variability and Atypical Phenotype-Genotype Correlations in Myopathies: Experience on A Cohort of 156 Families. Genes. 2021; 12(8):1199. https://doi.org/10.3390/genes12081199
Chicago/Turabian StyleJuntas Morales, Raul, Aurélien Perrin, Guilhem Solé, Delphine Lacourt, Henri Pegeot, Ulrike Walther-Louvier, Pascal Cintas, Claude Cances, Caroline Espil, Corinne Theze, and et al. 2021. "An Integrated Clinical-Biological Approach to Identify Interindividual Variability and Atypical Phenotype-Genotype Correlations in Myopathies: Experience on A Cohort of 156 Families" Genes 12, no. 8: 1199. https://doi.org/10.3390/genes12081199
APA StyleJuntas Morales, R., Perrin, A., Solé, G., Lacourt, D., Pegeot, H., Walther-Louvier, U., Cintas, P., Cances, C., Espil, C., Theze, C., Zenagui, R., Yauy, K., Cosset, E., Renard, D., Rigau, V., Maues de Paula, A., Uro-Coste, E., Arne-Bes, M.-C., Martin Négrier, M.-L., ... Cossée, M. (2021). An Integrated Clinical-Biological Approach to Identify Interindividual Variability and Atypical Phenotype-Genotype Correlations in Myopathies: Experience on A Cohort of 156 Families. Genes, 12(8), 1199. https://doi.org/10.3390/genes12081199