
Whole-Genome Differentially Hydroxymethylated DNA Regions among Twins Discordant for Cardiovascular Death

 ,
, 
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Study Samples
2.3. DNA Sample Collection
2.4. Genome-Wide Methylation (5mC) Measures
2.5. Measurement of Whole-Genome Hydroxymethylation (5hmC)
2.5.1. Genomic DNA Preparation
2.5.2. 5hmC Capture and Sequencing
2.6. Assessment of Covariates
2.7. Follow-up and Assessment of Endpoints
2.8. Statistical Analysis
2.8.1. Estimation of Peripheral Blood Leukocyte Composition
2.8.2. Identification of Signature Differentially Hydroxymethylated Regions
2.9. Bioinformatic Analysis
2.9.1. Bioinformatic Visualization
2.9.2. Functional Enrichment Analysis
2.9.3. DNA Motif Enrichment Analysis
3. Results
3.1. Characteristics of the Study Twin Pairs Discordant for Cardiovascular Death
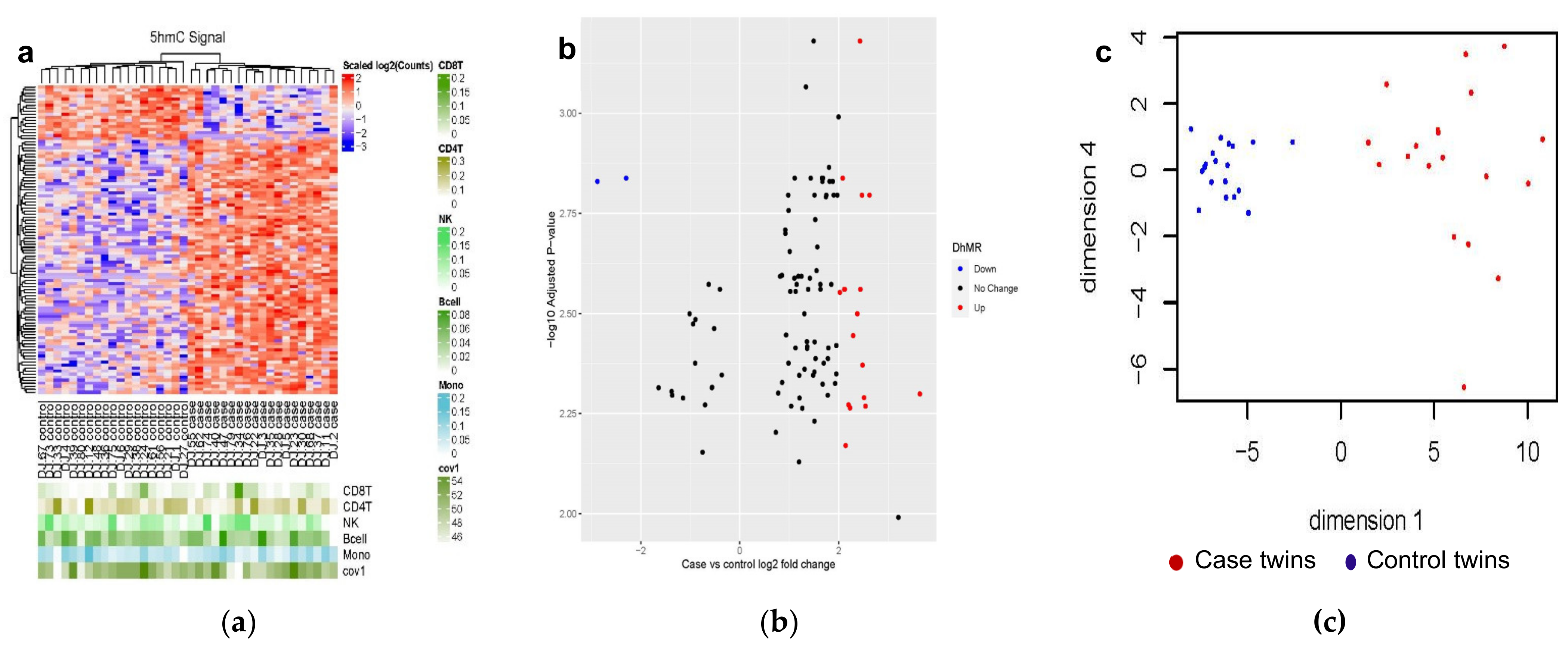
3.2. Differentially Hydroxymethylated Regions (DhMRs) from Monozygotic (MZ) Twin Pairs Discordant for Cardiovascular Death (CVD-dMZ)
3.2.1. Genetic Characteristics of the 102 DhMRs
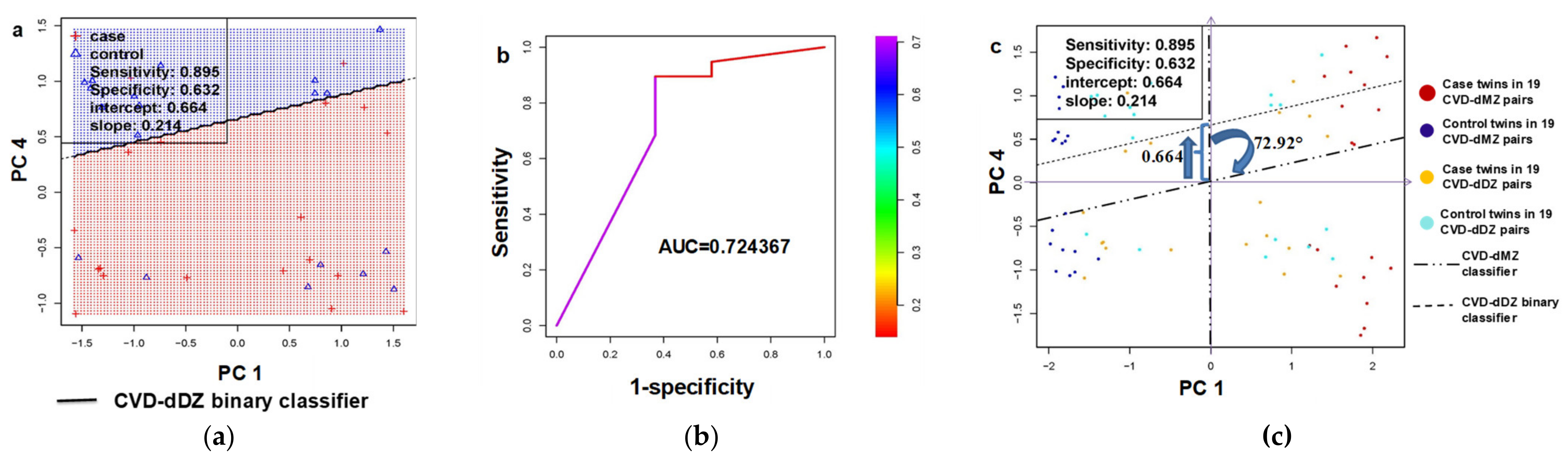
3.2.2. Generalizability Validation in Dizygotic (DZ) Twin Pairs Discordant for Cardiovascular Death (CVD-dDZ)
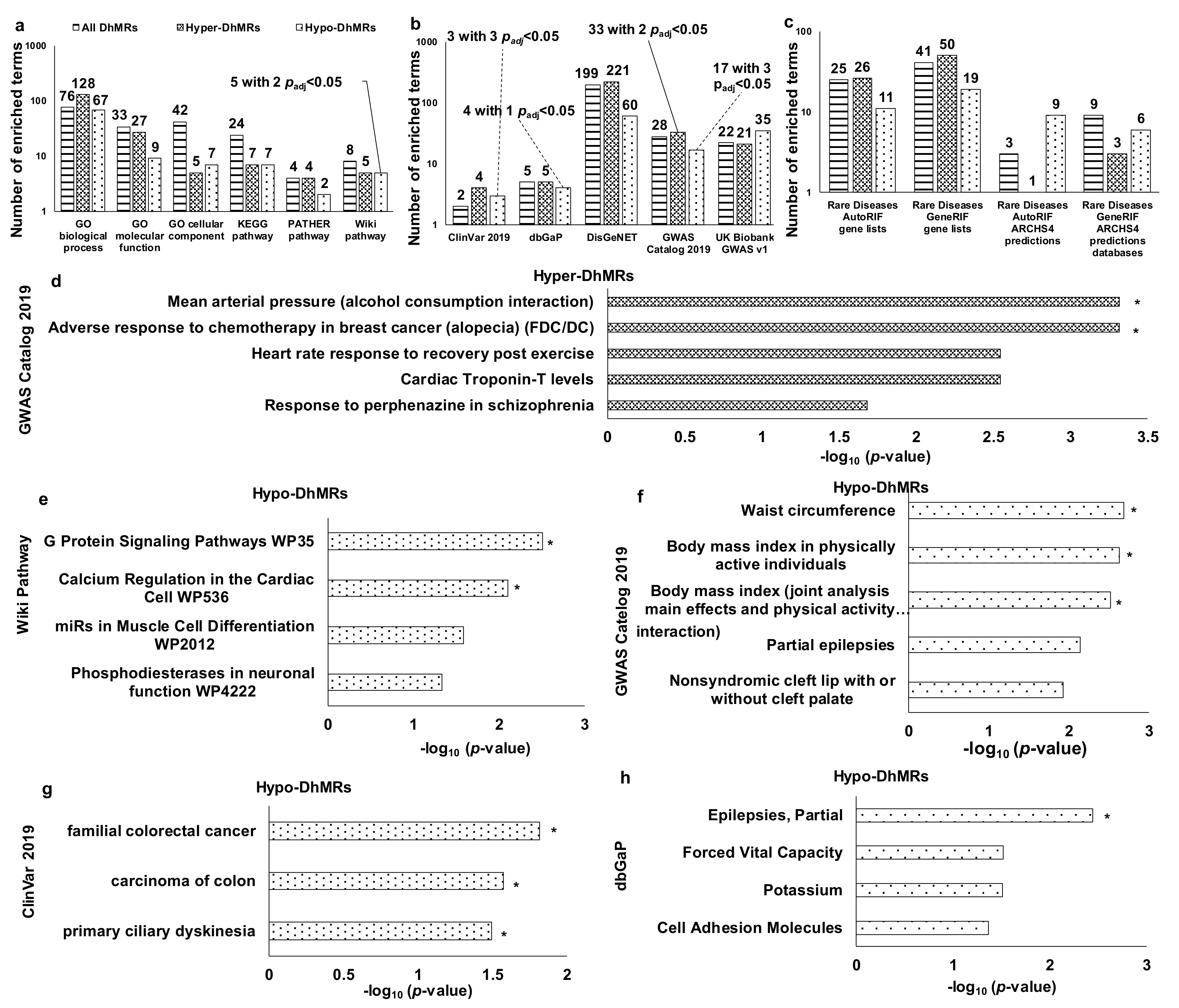
3.3. Functional Enrichment Analysis of DhMRs
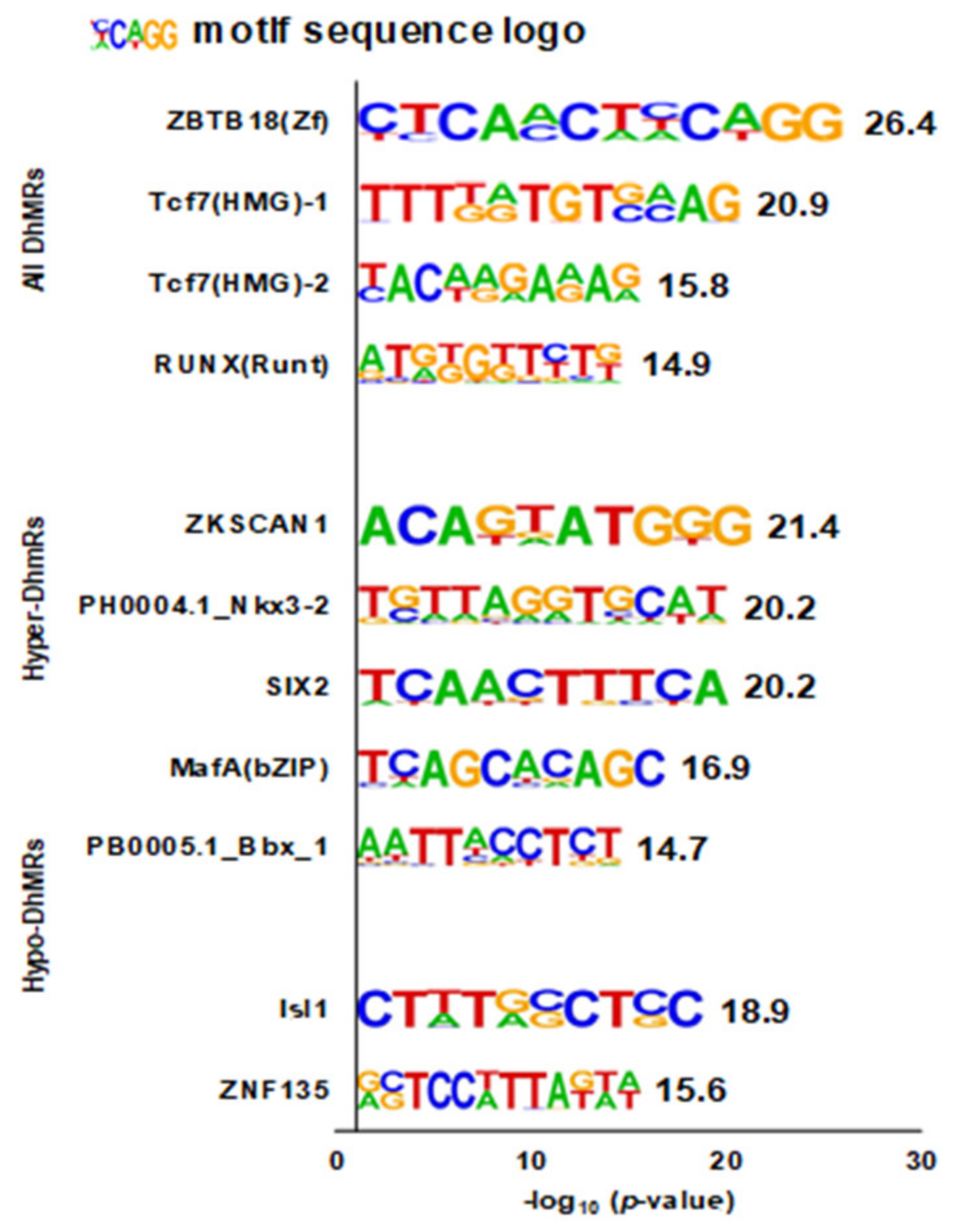
3.4. Enriched DNA Motifs
4. Discussion
4.1. Consistency with Prior Studies
4.2. Mechanisms
4.3. Limitations and Advantages
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abi Khalil, C. The emerging role of epigenetics in cardiovascular disease. Ther. Adv. Chronic Dis. 2014, 5, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Richa, R.; Sinha, R.P. Hydroxymethylation of DNA: An epigenetic marker. EXCLI J. 2014, 13, 592–610. [Google Scholar] [PubMed]
- Wang, H.; Lou, D.; Wang, Z. Crosstalk of Genetic Variants, Allele-Specific DNA Methylation, and Environmental Factors for Complex Disease Risk. Front. Genet. 2018, 9, 695. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Q.; Ali, I.; Tang, J.; Yang, W.C. New Insights into 5hmC DNA Modification: Generation, Distribution and Function. Front. Genet. 2017, 8, 100. [Google Scholar] [CrossRef]
- Delatte, B.; Jeschke, J.; Defrance, M.; Bachman, M.; Creppe, C.; Calonne, E.; Bizet, M.; Deplus, R.; Marroqui, L.; Libin, M.; et al. Genome-wide hydroxymethylcytosine pattern changes in response to oxidative stress. Sci. Rep. 2015, 5, 12714. [Google Scholar] [CrossRef]
- Tabish, A.M.; Arif, M.; Song, T.; Elbeck, Z.; Becker, R.C.; Knöll, R.; Sadayappan, S. Association of intronic DNA methylation and hydroxymethylation alterations in the epigenetic etiology of dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H168–H180. [Google Scholar] [CrossRef]
- Jiang, D.; Sun, M.; You, L.; Lu, K.; Gao, L.; Hu, C.; Wu, S.; Chang, G.; Tao, H.; Zhang, D. DNA methylation and hydroxymethylation are associated with the degree of coronary atherosclerosis in elderly patients with coronary heart disease. Life Sci. 2019, 224, 241–248. [Google Scholar] [CrossRef]
- Dong, C.; Chen, J.; Zheng, J.; Liang, Y.; Yu, T.; Liu, Y.; Gao, F.; Long, J.; Chen, H.; Zhu, Q.; et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic and predictive biomarkers for coronary artery disease. Clin. Epigenetics 2020, 12, 17. [Google Scholar] [CrossRef]
- Dai, J.; Krasnow, R.; Liu, L.; Sawada, S.; Reed, T. The association between postload plasma glucose levels and 38-year mortality risk of coronary heart disease: The prospective NHLBI Twin Study. PLoS ONE 2013, 8, e69332. [Google Scholar] [CrossRef]
- Bell, J.T.; Spector, T.D. A twin approach to unraveling epigenetics. Trends Genet. 2011, 27, 116–125. [Google Scholar] [CrossRef]
- Fabsitz, R.R.; Kalousdian, S.; Carmelli, D.; Robinette, D.; Christian, J.C. Characteristics of participants and nonparticipants in the NHLBI Twin Study. Acta Genet. Med. Gemellol. 1988, 37, 217–228. [Google Scholar] [CrossRef]
- Dai, J.; Mukamal, K.J.; Krasnow, R.E.; Swan, G.E.; Reed, T. Higher usual alcohol consumption was associated with a lower 41-y mortality risk from coronary artery disease in men independent of genetic and common environmental factors: The prospective NHLBI Twin Study. Am. J. Clin. Nutr. 2015, 102, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.; Carmelli, D.; Christian, J.C.; Selby, J.V.; Fabsitz, R.R. The NHLBI male veteran twin study data. Genet. Epidemiol. 1993, 10, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Ernster, V.L. Nested case-control studies. Prev. Med. 1994, 23, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.J.; Campen, D.; Hui, R.; Spence, M.; Cheetham, C.; Levy, G.; Shoor, S.; Ray, W.A. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: Nested case-control study. Lancet 2005, 365, 475–481. [Google Scholar] [CrossRef]
- Absher, D.M.; Li, X.; Waite, L.L.; Gibson, A.; Roberts, K.; Edberg, J.; Chatham, W.W.; Kimberly, R.P. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet. 2013, 9, e1003678. [Google Scholar] [CrossRef]
- Song, C.X.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.H.; Zhang, W.; Jian, X.; et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Han, J.W.; Kim, S.; Namburi, S.; Hermetz, K.; Kim, J.J.; Rudd, M.K.; et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 2011, 7, e1002154. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Shen, Y.; Park, B.; et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, E.; Lessov-Schlaggar, C.N.; Krasnow, R.E.; Swan, G.E. Nature versus nurture in gout: A twin study. Am. J. Med. 2012, 125, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Bhasin, J.M.; Hu, B.; Ting, A.H. MethylAction: Detecting differentially methylated regions that distinguish biological subtypes. Nucleic Acids Res. 2016, 44, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Irizarry, R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014, 15, R31. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Royal Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Mi, H.; Thomas, P. PANTHER pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Villaveces, J.M.; Koti, P.; Habermann, B.H. Tools for visualization and analysis of molecular networks, pathways, and -omics data. Adv. Appl. Bioinform. Chem. 2015, 8, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Mailman, M.D.; Feolo, M.; Jin, Y.; Kimura, M.; Tryka, K.; Bagoutdinov, R.; Hao, L.; Kiang, A.; Paschall, J.; Phan, L.; et al. The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 2007, 39, 1181–1186. [Google Scholar] [CrossRef]
- Pinero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutierrez-Sacristan, A.; Deu-Pons, J.; Centeno, E.; Garcia-Garcia, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2011, 39, D52–D57. [Google Scholar] [CrossRef]
- Jiang, D.; Wang, Y.; Chang, G.; Duan, Q.; You, L.; Sun, M.; Hu, C.; Gao, L.; Wu, S.; Tao, H.; et al. DNA hydroxymethylation combined with carotid plaques as a novel biomarker for coronary atherosclerosis. Aging 2019, 11, 3170–3181. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Gorlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.J.; Rao, A. TET Enzymes and 5hmC in Adaptive and Innate Immune Systems. Front. Immunol. 2019, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Tang, Z.H.; Ren, Z.; He, B.; Zeng, Y.; Liu, L.S.; Wang, Z.; Wei, D.H.; Zheng, X.L.; Jiang, Z.S. TET2 Protects against oxLDL-Induced HUVEC Dysfunction by Upregulating the CSE/H2S System. Front. Pharmacol. 2017, 8, 486. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, W.; Qu, K.; Lin, X.; Zeng, Z.; Chen, J.; Wei, D.; Wang, Z. TET2: A Novel Epigenetic Regulator and Potential Intervention Target for Atherosclerosis. DNA Cell Biol. 2018, 37, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chavez, L.; Chang, X.; Wang, X.; Pastor, W.A.; Kang, J.; Zepeda-Martinez, J.A.; Pape, U.J.; Jacobsen, S.E.; Peters, B.; et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1361–1366. [Google Scholar] [CrossRef]
- Ngo, T.T.; Yoo, J.; Dai, Q.; Zhang, Q.; He, C.; Aksimentiev, A.; Ha, T. Effects of cytosine modifications on DNA flexibility and nucleosome mechanical stability. Nat. Commun. 2016, 7, 10813. [Google Scholar] [CrossRef]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161–172. [Google Scholar] [CrossRef]
- Miao, L.; Yin, R.X.; Zhang, Q.H.; Hu, X.J.; Huang, F.; Chen, W.X.; Cao, X.L.; Wu, J.Z. Integrated DNA methylation and gene expression analysis in the pathogenesis of coronary artery disease. Aging 2019, 11, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Li, J.; Xiao, Y.; Lee, M.; Guo, L.; Han, W.; Li, T.; Hill, M.C.; Hong, T.; Mo, W.; et al. Tet inactivation disrupts YY1 binding and long-range chromatin interactions during embryonic heart development. Nat. Commun. 2019, 10, 4297. [Google Scholar] [CrossRef] [PubMed]
- Das, M.K.; Dai, H.-K. A survey of DNA motif finding algorithms. BMC Bioinform. 2007, 8 (Suppl. 7), S21. [Google Scholar] [CrossRef]
- Shrimankar, D. High performance computing approach for DNA motif discovery. CSI Trans. ICT 2019, 7, 295–297. [Google Scholar] [CrossRef]
- Boeva, V. Analysis of Genomic Sequence Motifs for Deciphering Transcription Factor Binding and Transcriptional Regulation in Eukaryotic Cells. Front. Genet. 2016, 7, 24. [Google Scholar] [CrossRef]
- Yokoyama, S.; Asahara, H. The myogenic transcriptional network. Cell. Mol. Life Sci. 2011, 68, 1843–1849. [Google Scholar] [CrossRef]
- Okado, H. Regulation of brain development and brain function by the transcriptional repressor RP58. Brain Res. 2019, 1705, 15–23. [Google Scholar] [CrossRef]
- Mahe, E.A.; Madigou, T.; Salbert, G. Reading cytosine modifications within chromatin. Transcription 2018, 9, 240–247. [Google Scholar] [CrossRef]
- Lercher, L.; McDonough, M.A.; El-Sagheer, A.H.; Thalhammer, A.; Kriaucionis, S.; Brown, T.; Schofield, C.J. Structural insights into how 5-hydroxymethylation influences transcription factor binding. Chem. Commun. 2014, 50, 1794–1796. [Google Scholar] [CrossRef]
- Yokoyama, S.; Ito, Y.; Ueno-Kudoh, H.; Shimizu, H.; Uchibe, K.; Albini, S.; Mitsuoka, K.; Miyaki, S.; Kiso, M.; Nagai, A.; et al. A systems approach reveals that the myogenesis genome network is regulated by the transcriptional repressor RP58. Dev. Cell 2009, 17, 836–848. [Google Scholar] [CrossRef]
- Seyerle, A.A.; Lin, H.J.; Gogarten, S.M.; Stilp, A.; Mendez Giraldez, R.; Soliman, E.; Baldassari, A.; Graff, M.; Heckbert, S.; Kerr, K.F.; et al. Genome-wide association study of PR interval in Hispanics/Latinos identifies novel locus at ID2. Heart 2018, 104, 904–911. [Google Scholar] [CrossRef]
- Doran, A.C.; Lehtinen, A.B.; Meller, N.; Lipinski, M.J.; Slayton, R.P.; Oldham, S.N.; Skaflen, M.D.; Yeboah, J.; Rich, S.S.; Bowden, D.W.; et al. Id3 is a novel atheroprotective factor containing a functionally significant single-nucleotide polymorphism associated with intima-media thickness in humans. Circ. Res. 2010, 106, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Rao, S.; Shen, G.Q.; Li, L.; Moliterno, D.J.; Newby, L.K.; Rogers, W.J.; Cannata, R.; Zirzow, E.; Elston, R.C.; et al. Premature myocardial infarction novel susceptibility locus on chromosome 1P34-36 identified by genomewide linkage analysis. Am. J. Hum. Genet. 2004, 74, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Manichaikul, A.; Rich, S.S.; Perry, H.; Yeboah, J.; Law, M.; Davis, M.; Parker, M.; Ragosta, M.; Connelly, J.J.; McNamara, C.A.; et al. A functionally significant polymorphism in ID3 is associated with human coronary pathology. PLoS ONE 2014, 9, e90222. [Google Scholar] [CrossRef] [PubMed]
- Shendre, A.; Irvin, M.R.; Wiener, H.; Zhi, D.; Limdi, N.A.; Overton, E.T.; Shrestha, S. Local Ancestry and Clinical Cardiovascular Events Among African Americans From the Atherosclerosis Risk in Communities Study. J. Am. Heart Assoc. 2017, 6, e004739. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, D.; Tsuchiya, N.; Yamaguchi, A.; Okaji, Y.; Tsuno, N.H.; Kobata, T.; Takahashi, K.; Tokunaga, K. Crucial role of inhibitor of DNA binding/differentiation in the vascular endothelial growth factor-induced activation and angiogenic processes of human endothelial cells. J. Immunol. 2004, 173, 5801–5809. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, X.; Morrell, N.W. Id proteins in the vasculature: From molecular biology to cardiopulmonary medicine. Cardiovasc. Res. 2014, 104, 388–398. [Google Scholar] [CrossRef]
- Yang, J.; Li, X.; Li, Y.; Southwood, M.; Ye, L.; Long, L.; Al-Lamki, R.S.; Morrell, N.W. Id proteins are critical downstream effectors of BMP signaling in human pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L312–L321. [Google Scholar] [CrossRef] [PubMed]
- Camare, C.; Pucelle, M.; Negre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Miura, T.; Kawana, H.; Nonaka, K. Twinning in New England in the 17th–19th centuries. Acta Genet. Med. Gemellol. 1987, 36, 355–364. [Google Scholar] [CrossRef]
- Fellman, J.; Eriksson, A.W. The convergence of the regional twinning rates in Sweden, 1751–1960. Twin Res. Hum. Genet. 2005, 8, 163–172. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Strandskov, H.H.; Edelen, E.W. Monozygotic and dizygotic twin birth frequencies in the total, the “white” and the “colored” U.S. populations. Genetics 1946, 31, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Live Births and Birth Rates by Year. Infoplease© 2021–2012 Pearson Education. 2007. Available online: http://www.infoplease.com/ipa/A0005067.html (accessed on 27 July 2021).
- Mahmood, S.S.; Levy, D.; Vasan, R.S.; Wang, T.J. The Framingham Heart Study and the epidemiology of cardiovascular disease: A historical perspective. Lancet 2014, 383, 999–1008. [Google Scholar] [CrossRef]
- Babyak, M.A. What you see may not be what you get: A brief, nontechnical introduction to overfitting in regression-type models. Psychosom. Med. 2004, 66, 411–421. [Google Scholar]
- Houseman, E.A.; Kim, S.; Kelsey, K.T.; Wiencke, J.K. DNA Methylation in Whole Blood: Uses and Challenges. Curr. Environ. Health Rep. 2015, 2, 145–154. [Google Scholar] [CrossRef]
- Wu, S.-H.; Neale, M.C.; Acton, A.J., Jr.; Considine, R.V.; Krasnow, R.E.; Reed, T.; Dai, J. Genetic and environmental influences on the prospective correlation between systemic inflammation and coronary heart disease death in male twins. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2168–2174. [Google Scholar] [CrossRef]
- Kim, R.S. Analysis of Nested Case-Control Study Designs: Revisiting the Inverse Probability Weighting Method. Commun. Stat. Appl. Methods 2013, 20, 455–466. [Google Scholar] [CrossRef]
- Wang, M.H.; Shugart, Y.Y.; Cole, S.R.; Platz, E.A. A simulation study of control sampling methods for nested case-control studies of genetic and molecular biomarkers and prostate cancer progression. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 706–711. [Google Scholar] [CrossRef]
- Kruglyak, L.; Nickerson, D.A. Variation is the spice of life. Nat. Genet. 2001, 27, 234–236. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotation 1 | Total DhMRs | Hyper-DhMRs | Hypo-DhMRs |
|---|---|---|---|
| n | 102 | 84 | 18 |
| Exons1 | 2 (2.0 3) | 2 (2.3 3) | 0 |
| ncRNA2 | 1 (50 4) | 1 (50 4) | 0 |
| protein-coding2 | 1 (50 4) | 1 (50 4) | 0 |
| Pseudo2 | 0 | 0 | 0 |
| snoRNA2 | 0 | 0 | 0 |
| Intergenic regions1 | 47 (46 3) | 42 (50 3) | 5 (28 3) |
| ncRNA | 14 (30 4) | 13 (31 4) | 1 (20 4) |
| protein-coding | 30 (64 4) | 26 (62 4) | 4 (80 4) |
| pseudo | 3 (6 4) | 3 (7 4) | 0 |
| snoRNA | 0 | 0 | 0 |
| Introns1 | 49 (48 3) | 37 (44 3) | 12 (67 3) |
| ncRNA | 11 (22 4) | 10 (27 4) | 1 (8 4) |
| protein-coding | 35 (71 4) | 25 (68 4) | 10 (83 4) |
| pseudo | 3 (6 4) | 1 (3 4) | 1 (8 4) |
| snoRNA | 1 (2 4) | 1 (3 4) | 0 |
| TSS1 | 2 (2.0 3) | 1 (1.2 3) | 1 (5.6 3) |
| ncRNA | 0 | 0 | 0 |
| protein-coding | 1 (50 4) | 0 | 1 (100 4) |
| pseudo | 1 (50 4) | 1 (100 4) | 0 |
| snoRNA | 0 | 0 | 0 |
| TTS 1 | 2 (2.0 3) | 2 (2.3 3) | 0 |
| ncRNA | 0 | 0 | 0 |
| protein-coding | 2 (100 4) | 2 (100 4) | 0 |
| pseudo | 0 | 0 | 0 |
| snoRNA | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, J.; Leung, M.; Guan, W.; Guo, H.-T.; Krasnow, R.E.; Wang, T.J.; El-Rifai, W.; Zhao, Z.; Reed, T. Whole-Genome Differentially Hydroxymethylated DNA Regions among Twins Discordant for Cardiovascular Death. Genes 2021, 12, 1183. https://doi.org/10.3390/genes12081183
Dai J, Leung M, Guan W, Guo H-T, Krasnow RE, Wang TJ, El-Rifai W, Zhao Z, Reed T. Whole-Genome Differentially Hydroxymethylated DNA Regions among Twins Discordant for Cardiovascular Death. Genes. 2021; 12(8):1183. https://doi.org/10.3390/genes12081183
Chicago/Turabian StyleDai, Jun, Ming Leung, Weihua Guan, Han-Tian Guo, Ruth E. Krasnow, Thomas J. Wang, Wael El-Rifai, Zhongming Zhao, and Terry Reed. 2021. "Whole-Genome Differentially Hydroxymethylated DNA Regions among Twins Discordant for Cardiovascular Death" Genes 12, no. 8: 1183. https://doi.org/10.3390/genes12081183
APA StyleDai, J., Leung, M., Guan, W., Guo, H.-T., Krasnow, R. E., Wang, T. J., El-Rifai, W., Zhao, Z., & Reed, T. (2021). Whole-Genome Differentially Hydroxymethylated DNA Regions among Twins Discordant for Cardiovascular Death. Genes, 12(8), 1183. https://doi.org/10.3390/genes12081183