
Mitochondrial Genomes from Two Specialized Subfamilies of Reduviidae (Insecta: Hemiptera) Reveal Novel Gene Rearrangements of True Bugs


Abstract
:1. Introduction
2. Materials and Methods
2.1. Assassin Bugs Collection and DNA Extraction
2.2. Genome Sequencing, Assembly and Analyses
2.3. Phylogenetic Analyses
3. Results and discussion
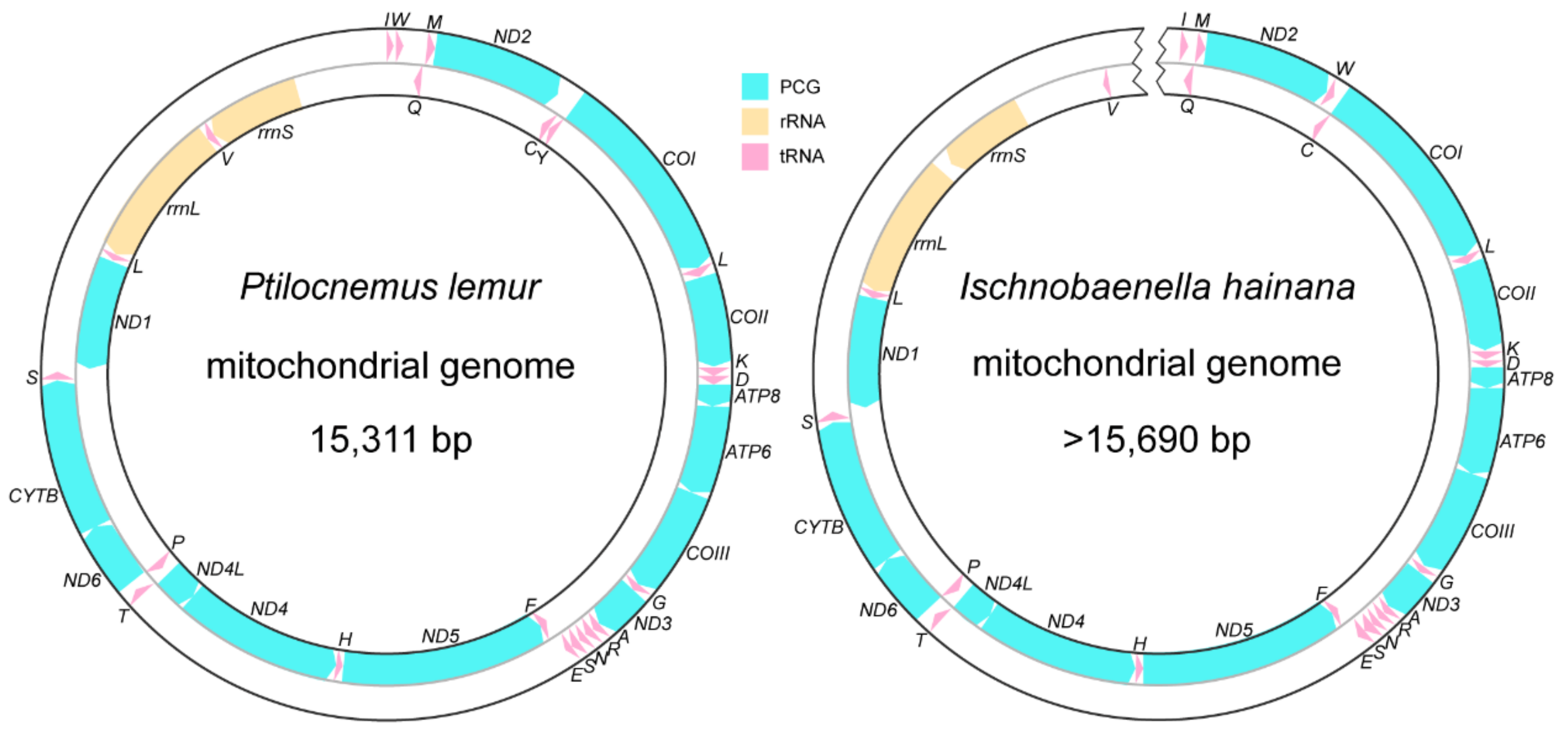
3.1. The General Features of Assassin Bugs Mitogenomes
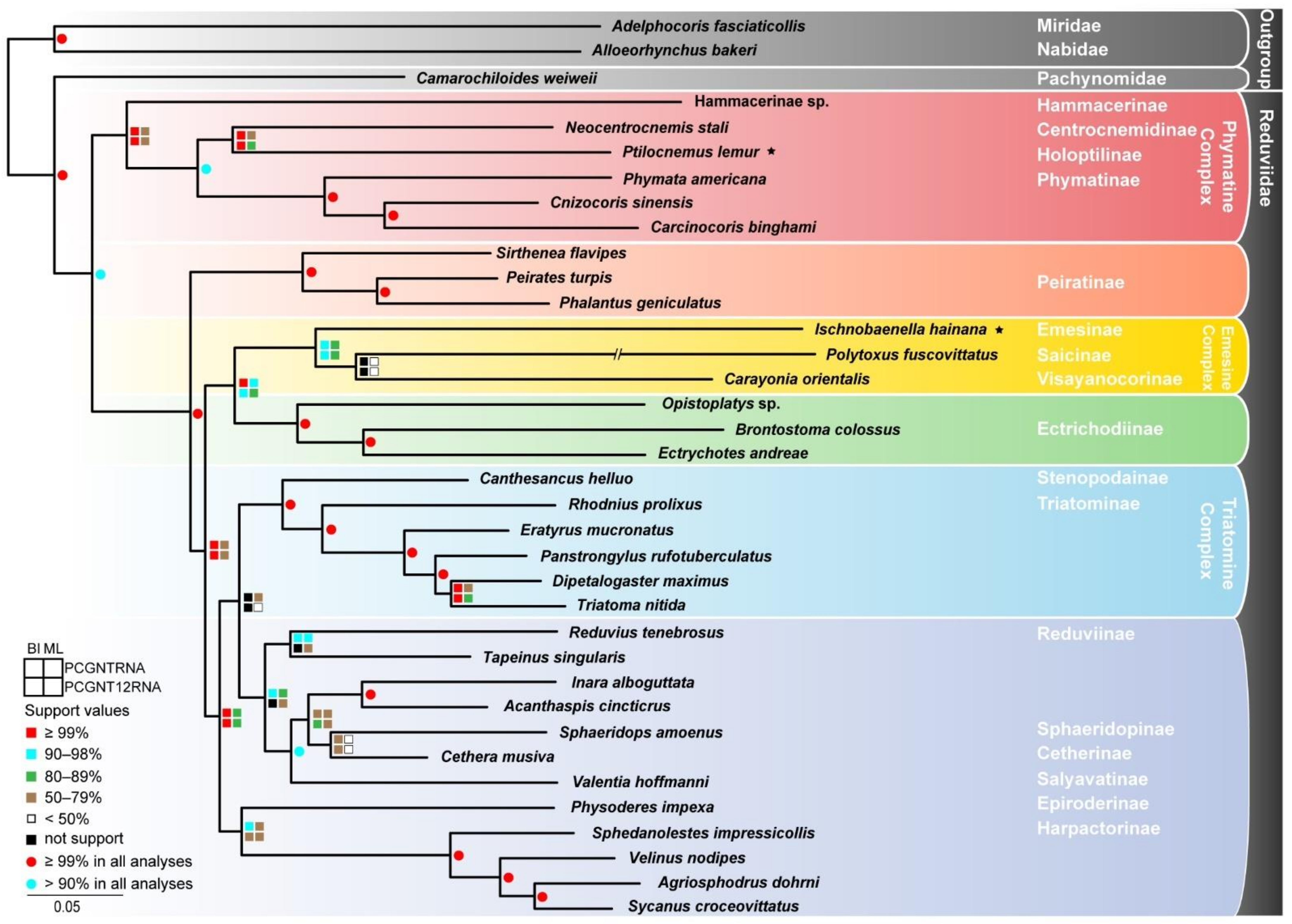
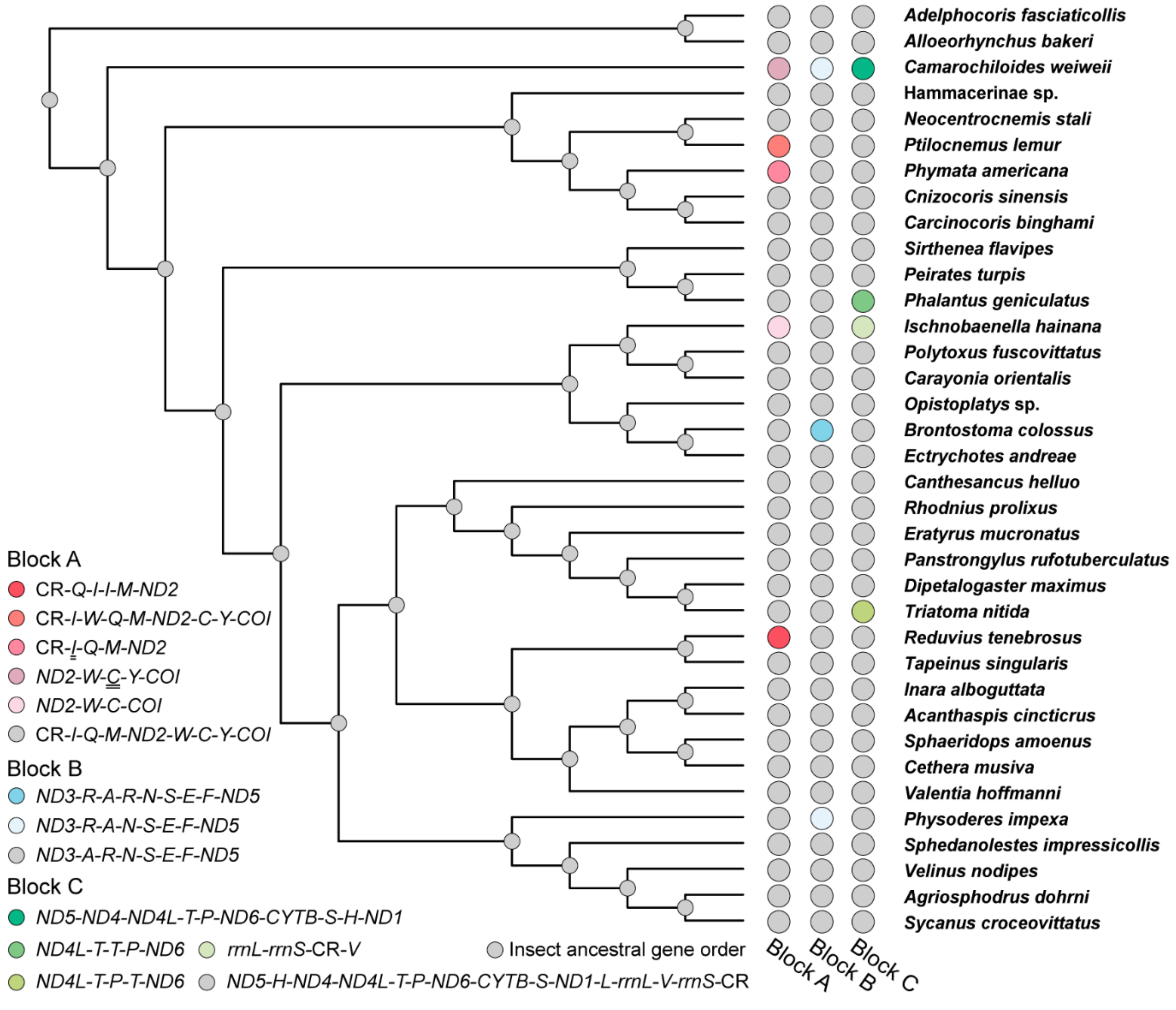
3.2. Phylogenetic Analyses
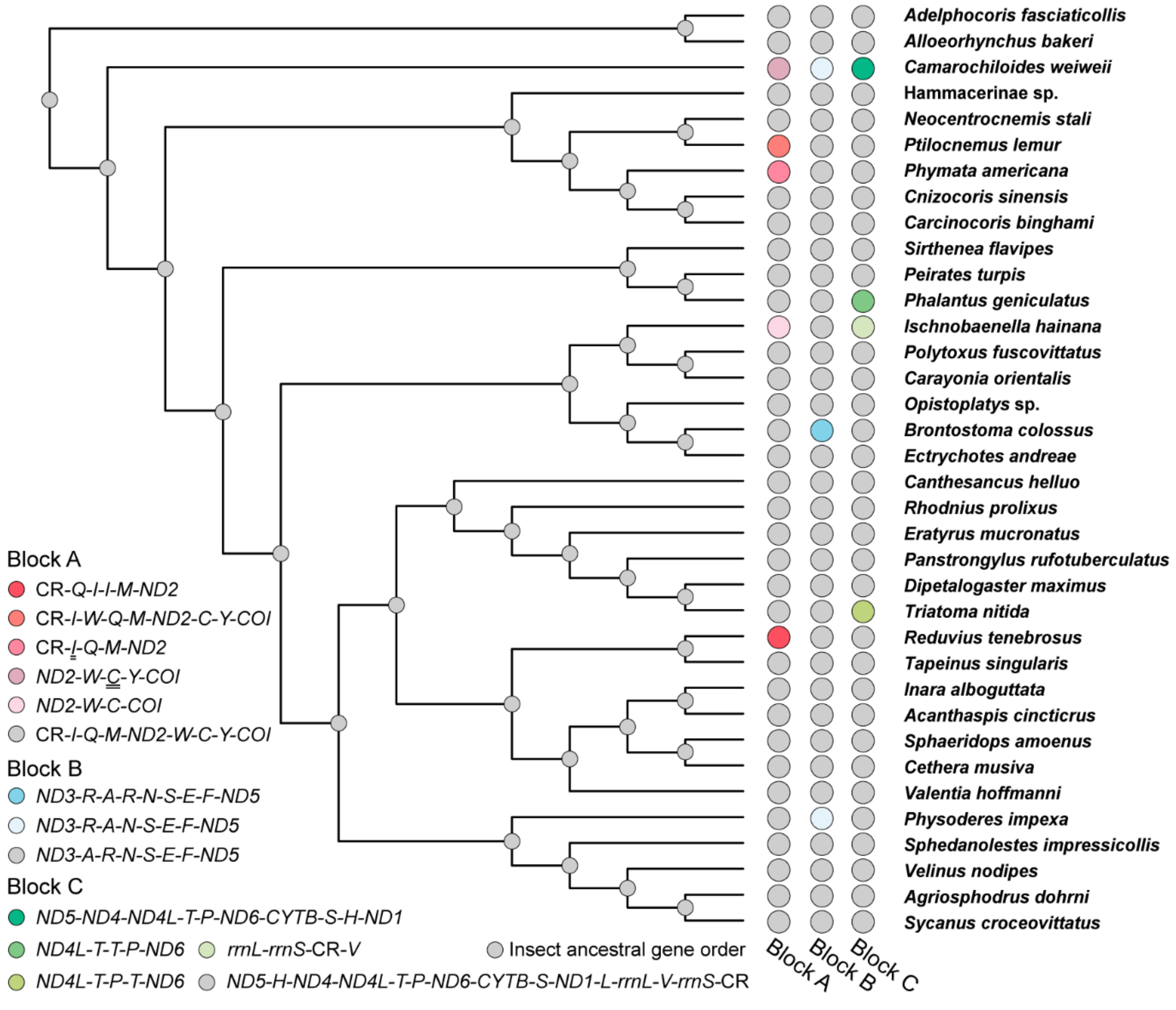
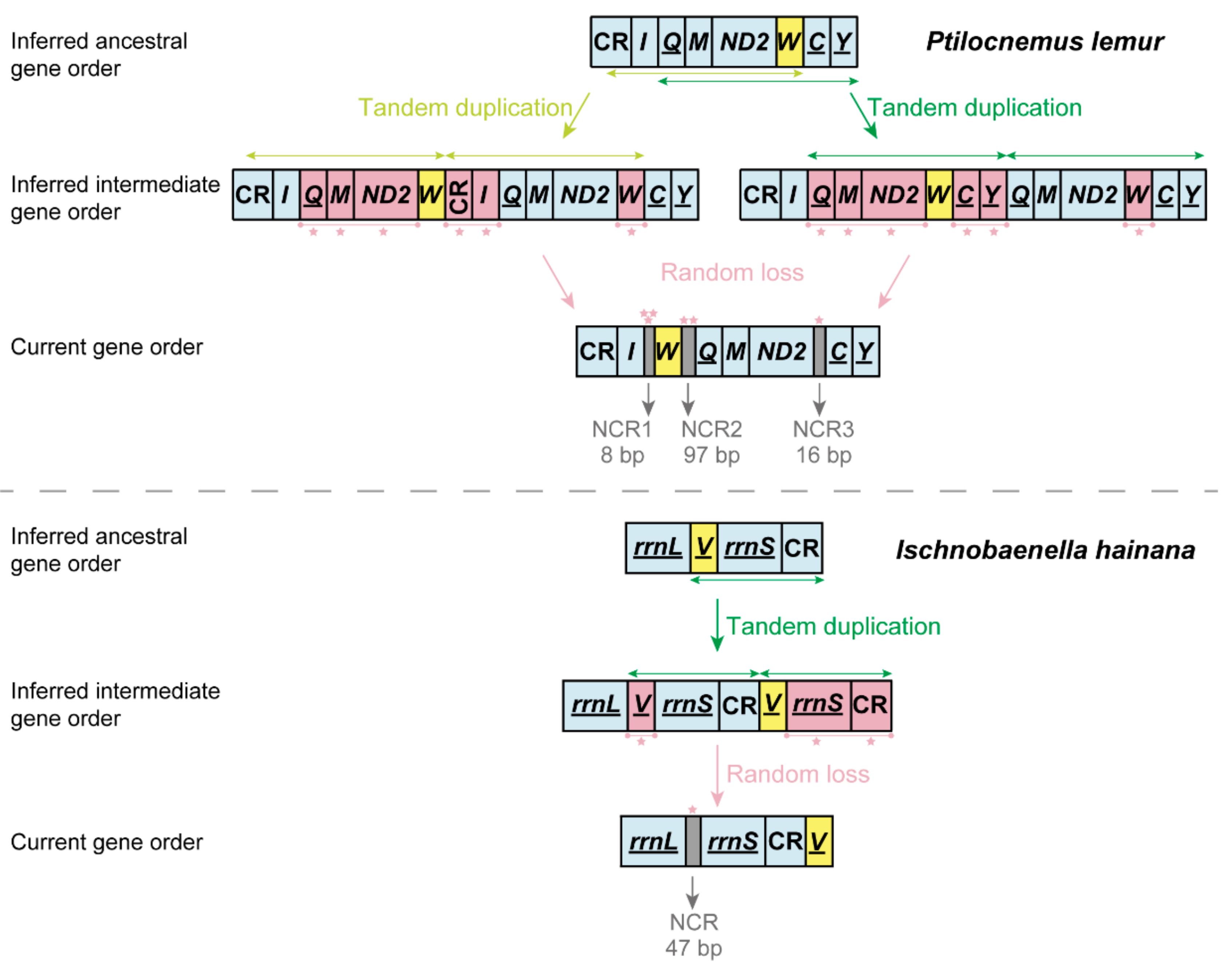
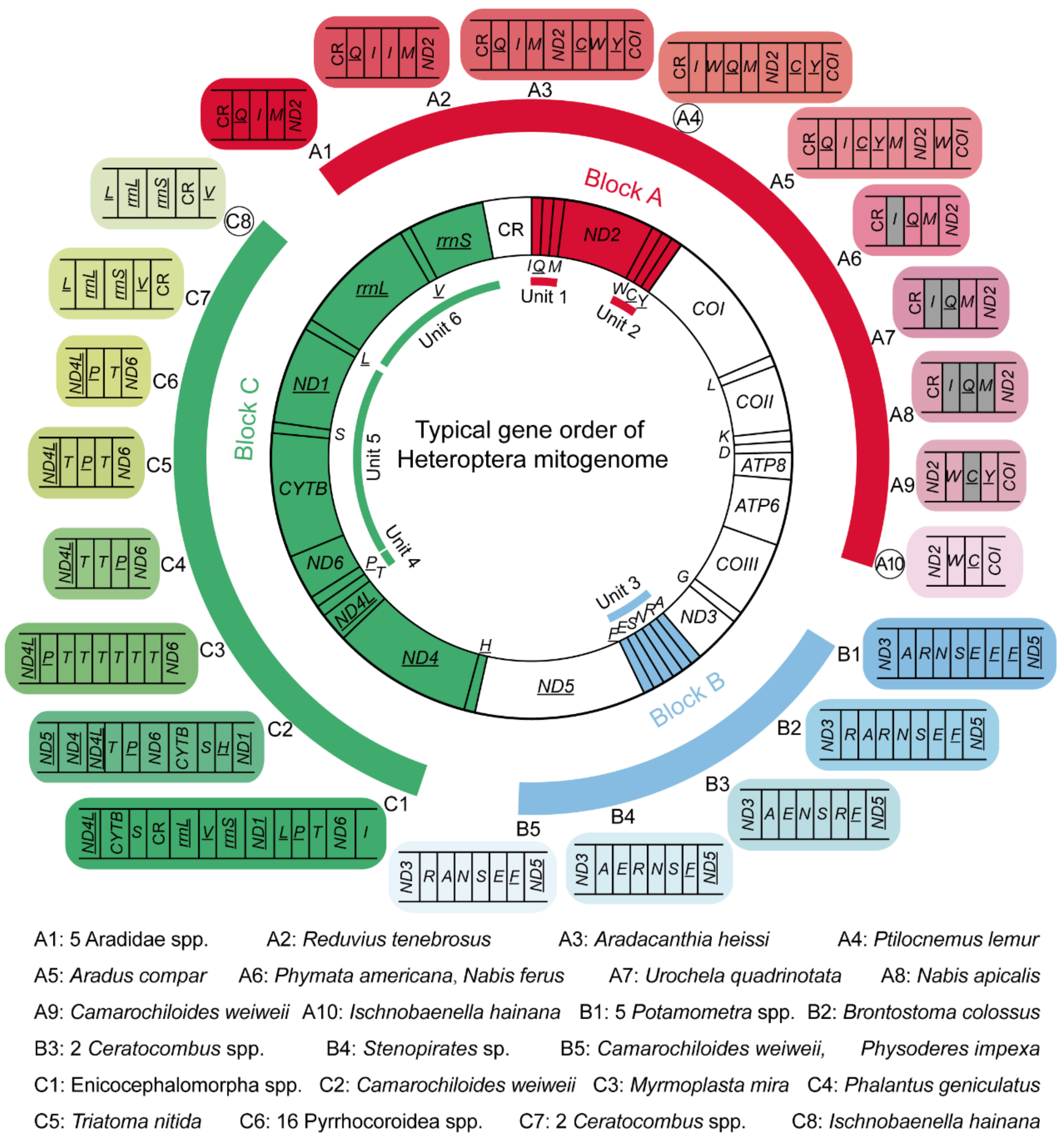
3.3. Gene Rearrangement in Heteroptera Mitogenomes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newmeyer, D.D.; Ferguson-Miller, S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell 2003, 112, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, S.; Hadrys, H. A comparative analysis of complete mitochondrial genomes among Hexapoda. Mol. Phylogenet. Evol. 2013, 69, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. Phylogenetic position of the Pentastomida and (pan)crustacean relationships. Proc. R. Soc. Lond. B 2004, 271, 537–544. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Johnson, K.P.; Sweet, A.D.; Yao, I.; Ferreira, R.L.; Cameron, S.L. Mitochondrial phylogenomics and genome rearrangements in the barklice (Insecta: Psocodea). Mol. Phylogenet. Evol. 2018, 119, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Li, H.; Liu, G.-H.; Wang, W.; James, P.; Colwell, D.D.; Tran, A.; Gong, S.; Cai, W.; Shao, R. Mitochondrial genome fragmentation unites the parasitic lice of eutherian mammals. Syst. Biol. 2019, 68, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Dickey, A.M.; Kumar, V.; Morgan, J.K.; Jara-Cavieres, A.; Shatters, R.G.J.; McKenzie, C.L.; Osborne, L.S. A novel mitochondrial genome architecture in thrips (Insecta: Thysanoptera): Extreme size asymmetry among chromosomes and possible recent control region duplication. BMC Genom. 2015, 16, 439. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, K.; Chakraborty, R.; Cameron, S.L.; Sweet, A.D.; Chandra, K.; Kumar, V. Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta). Sci. Rep. 2020, 10, 695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.; Zhu, J.C.; Zheng, B.Y.; Wei, S.J.; Sharkey, M.; Chen, X.X.; Vogler, A.P. Mitochondrial phylogenomics of the Hymenoptera. Mol. Phylogenet. Evol. 2019, 131, 8–18. [Google Scholar] [CrossRef]
- Mao, M.; Gibson, T.; Dowton, M. Evolutionary dynamics of the mitochondrial genome in the Evaniomorpha (Hymenoptera)—A group with an intermediate rate of gene rearrangement. Genome Biol. Evol. 2014, 6, 1862–1874. [Google Scholar] [CrossRef]
- Thao, M.L.; Baumann, L.; Baumann, P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha). BMC Evol. Biol. 2004, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, Q.; Lu, C.; Deng, J.; Huang, X. The first complete mitochondrial genome of Lachninae species and comparative genomics provide new insights into the evolution of gene rearrangement and the repeat region. Insects 2021, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Huang, X.; Deng, J. The challenge of Coccidae (Hemiptera: Coccoidea) mitochondrial genomes: The case of Saissetia coffeae with novel truncated tRNAs and gene rearrangements. Int. J. Biol. Macromol. 2020, 158, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.J.; Zhu, W.C.; Xia, R.; Zhang, Y.K.; Ding, X.L.; Liu, J.; Chen, D.S.; Yu, D.; Hong, X.Y. The complete mitochondrial genomes of two rice planthoppers, Nilaparvata lugens and Laodelphax striatellus: Conserved genome rearrangement in Delphacidae and discovery of new characteristics of atp8 and tRNA genes. BMC Genom. 2013, 14, 417. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, H.; Shi, A.; Štys, P.; Zhou, X.; Cai, W. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS ONE 2012, 7, e29419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Li, H.; Song, F.; Cai, Y.; Wang, J.; Liu, J.; Cai, W. Duplication and remolding of tRNA genes in the mitochondrial genome of Reduvius tenebrosus (Hemiptera: Reduviidae). Int. J. Mol. Sci. 2016, 17, 951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Liu, Y.; Wilson, J.J.; Chen, Z.; Song, F.; Cai, W.; Li, H. Mitochondrial genome of Phalantus geniculatus (Hemiptera: Reduviidae): trnT duplication and phylogenetic implications. Int. J. Biol. Macromol. 2019, 129, 110–115. [Google Scholar] [CrossRef]
- Kocher, A.; Kamilari, M.; Lhuillier, E.; Coissac, E.; Péneau, J.; Chave, J.; Murienne, J. Shotgun assembly of the assassin bug Brontostoma colossus mitochondrial genome (Heteroptera, Reduviidae). Gene 2014, 552, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, Y.; Wu, Y.; Song, F.; Cai, W.; Li, H. Novel tRNA gene rearrangements in the mitochondrial genome of Camarochiloides weiweii (Hemiptera: Pachynomidae). Int. J. Biol. Macromol. 2020, 165, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.; Hu, L.; Bai, X.; Dai, X.; Chang, J.; Guilbert, E.; Cai, W. The complete mitochondrial genome of the flat bug Aradacanthia heissi (Hemiptera: Aradidae). Zootaxa 2012, 3238, 23–38. [Google Scholar] [CrossRef]
- Hua, J.; Li, M.; Dong, P.; Cui, Y.; Xie, Q.; Bu, W. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genom. 2008, 9, 610. [Google Scholar] [CrossRef] [Green Version]
- Henry, T.J. Biodiversity of Heteroptera. In Insect Biodiversity: Science and Society, 2nd ed.; Foottit, R.G., Adler, P.H., Eds.; Wiley-Blackwell: Oxford, UK, 2017; pp. 279–335. [Google Scholar]
- Schuh, R.T.; Weirauch, C. True Bugs of the World (Hemiptera: Heteroptera): Classification and Natural History, 2nd ed.; Siri Scientific Press: Rochdale, UK, 2020; pp. 339–357. [Google Scholar]
- Weirauch, C.; Bérenger, J.M.; Berniker, L.; Forero, D.; Forthman, M.; Frankenberg, S.; Freedman, A.; Gordon, E.; Hoey-Chamberlain, R.; Hwang, W.S.; et al. An illustrated identification key to assassin bug subfamilies and tribes (Hemiptera: Reduviidae). Can. J. Arthropod Identific. 2014, 26, 1–115. [Google Scholar]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Gao, C.; Cui, Y.; Xie, Q.; Bu, W. The complete mitochondrial genome of the stalk-eyed bug Chauliops fallax Scott, and the monophyly of Malcidae (Hemiptera: Heteroptera). PLoS ONE 2013, 8, e55381. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Wang, Y.; Rédei, D.; Xie, Q.; Bu, W. Secondary structure models of 18S and 28S rRNAs of the true bugs based on complete rDNA sequences of Eurydema maracandica Oshanin, 1871 (Heteroptera, Pentatomidae). Zookeys 2013, 319, 363–377. [Google Scholar]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. Version 3.61. 2019. Available online: http://mesquiteproject.org (accessed on 1 July 2021).
- Zhao, Y.; Jiang, M.; Wu, Y.; Song, F.; Cai, W.; Li, H. Mitochondrial genomes of three kissing bugs (Reduviidae: Triatominae) and their phylogenetic implications. Int. J. Biol. Macromol. 2019, 134, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leavengood Jr, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. Lond. B 2017, 284, 20171223. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Song, F.; Shi, A.; Zhou, X.; Cai, W. Comparative mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS ONE 2012, 7, e45925. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, C. Cladistic analysis of Reduviidae (Heteroptera: Cimicomorpha) based on morphological characters. Syst. Entomol. 2008, 33, 229–274. [Google Scholar] [CrossRef]
- Weirauch, C.; Munro, J. Molecular phylogeny of the assassin bugs (Hemiptera: Reduviidae), based on mitochondrial and nuclear ribosomal genes. Mol. Phylogenet. Evol. 2009, 53, 287–299. [Google Scholar] [CrossRef]
- Zhang, J.; Weirauch, C.; Zhang, G.; Forero, D. Molecular phylogeny of Harpactorinae and Bactrodinae uncovers complex evolution of sticky trap predation in assassin bugs (Heteroptera: Reduviidae). Cladistics 2016, 32, 538–554. [Google Scholar] [CrossRef]
- Masonick, P.; Michael, A.; Frankenberg, S.; Rabitsch, W.; Weirauch, C. Molecular phylogenetics and biogeography of the ambush bugs (Hemiptera: Reduviidae: Phymatinae). Mol. Phylogenet. Evol. 2017, 114, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.S.; Weirauch, C. Evolutionary history of assassin bugs (Insecta: Hemiptera: Reduviidae): Insights from divergence dating and ancestral state reconstruction. PLoS ONE 2012, 7, e45523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gordon, E.R.L.; Forthman, M.; Hwang, W.S.; Walden, K.; Swanson, D.R.; Johnson, K.P.; Meier, R.; Weirauch, C. Evolution of the assassin’s arms: Insights from a phylogeny of combined transcriptomic and ribosomal DNA data (Heteroptera: Reduvioidea). Sci. Rep. 2016, 6, 22177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera-Uribe, M.; Meza-Lázaro, R.N.; Kieran, T.J.; Ibarra-Cerdeña, C.N.; Zaldívar-Riverón, A. Phylogeny of the North-Central American clade of blood-sucking reduviid bugs of the tribe Triatomini (Hemiptera: Triatominae) based on the mitochondrial genome. Infect. Genet. Evol. 2020, 84, 104373. [Google Scholar] [CrossRef] [PubMed]
- Macey, J.R.; Larson, A.; Ananjeva, N.B.; Fang, Z.; Papenfuss, T.J. Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol. Biol. Evol. 1997, 14, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In Comparative Genomics: Empirical and Analytical Approaches to Gene Order Dynamics, Map Alignment and the Evolution of Gene Families; Sankoff, D., Nadeau, J.H., Eds.; Computational Biology Series; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; Volume 1, pp. 133–147. [Google Scholar]
- Castro, L.R.; Austin, A.D.; Dowton, M. Contrasting rates of mitochondrial molecular evolution in parasitic Diptera and Hymenoptera. Mol. Biol. Evol. 2002, 19, 1100–1113. [Google Scholar] [CrossRef]
- Dowton, M.; Austin, A.D. Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol. Biol. Evol. 1999, 16, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunt, D.H.; Hyman, B.C. Animal mitochondrial DNA recombination. Nature 1997, 387, 247. [Google Scholar] [CrossRef]
- Chen, S.-C.; Wang, X.-Q.; Li, P.-W.; Hu, X.; Wang, J.-J.; Peng, P. The complete mitochondrial genome of Aleurocanthus camelliae: Insights into gene arrangement and genome organization within the family Aleyrodidae. Int. J. Mol. Sci. 2016, 17, 1843. [Google Scholar] [CrossRef] [Green Version]
- Le, T.H.; Blair, D.; McManus, D.P. Mitochondrial genomes of parasitic flatworms. Trends Parasitol. 2002, 18, 206–213. [Google Scholar] [CrossRef]
- Miyamoto, H.; Machida, R.J.; Nishida, S. Complete mitochondrial genome sequences of the three pelagic chaetognaths Sagitta nagae, Sagitta decipiens and Sagitta enflata. Comp. Biochem. Physiol. Part D Genom. Proteom. 2010, 5, 65–72. [Google Scholar] [CrossRef]
- Bhattacharyya, S.N.; Adhya, S. The complexity of mitochondrial tRNA import. RNA Biol. 2004, 1, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Börner, G.V.; Mörl, M.; Janke, A.; Pääbo, S. RNA editing changes the identity of a mitochondrial tRNA in marsupials. EMBO J. 1996, 15, 5949–5957. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, H.; Shao, R.; Shi, A.; Bai, X.; Zheng, X.; Heiss, E.; Cai, W. Rearrangement of mitochondrial tRNA genes in flat bugs (Hemiptera: Aradidae). Sci. Rep. 2016, 6, 25725. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infraorder | Family (Species Number) | Rearranged Gene Order | Potential Synapomorphy | Rearrangement Type |
|---|---|---|---|---|
| Enicocephalomorpha | Aenictopecheidae (1) | ND4L-CYTB-S-CR-rrnL-V-rrnS-ND1-L-P-T-ND6-I | infraorder | gene translocation |
| Enicocephalidae (4) | ||||
| Enicocephalidae (1) | A-E-R-N-S-F-//-ND4L-CYTB-S-CR-rrnL-V-rrnS-ND1-L-P-T-ND6-I | / | gene translocation | |
| Dipsocoromorpha | Ceratocombidae (2) | A-E-N-S-R-F-//-rrnL-rrnS-V-CR | genus | gene translocation |
| Gerromorpha | Gerridae (5) | A-R-N-S-E-F-F | genus | gene duplication |
| Cimicomorpha | Nabidae (1) | CR-I-Q-M-ND2 | / | gene loss |
| Nabidae (1) | CR-I-Q-M-ND2 | / | gene loss | |
| Pachynomidae (1) | ND2-W-C-Y-//-R-A-N-S-E-F-//-CYTB-S-H-ND1 | / | gene loss, gene translocation | |
| Reduviidae (1) | CR-Q-I-I-M-ND2 | / | gene duplication, gene translocation | |
| Reduviidae (1) | CR-I-W-Q-M-ND2-C-Y-COI | / | gene translocation | |
| Reduviidae (1) | ND2-W-C-COI-//-rrnL-rrnS-CR-V | / | gene translocation | |
| Reduviidae (1) | CR-I-Q-M-ND2 | / | gene loss | |
| Reduviidae (1) | R-A-R-N-S-E-F | / | gene duplication, gene translocation | |
| Reduviidae (1) | R-A-N-S-E-F | / | gene translocation | |
| Reduviidae (1) | ND4L-T-T-P-ND6 | / | gene duplication | |
| Reduviidae (1) | ND4L-T-P-T-ND6 | / | gene duplication, gene translocation | |
| Pentatomomorpha | Aradidae (5) | CR-Q-I-M-ND2 | family | gene translocation |
| Aradidae (1) | CR-Q-I-M-ND2-C-W-Y-COI | / | gene translocation | |
| Aradidae (1) | CR-Q-I-C-Y-M-ND2-W-COI | / | gene translocation | |
| Largidae (4) | ND4L-P-T-ND6 | superfamily | gene translocation | |
| Pyrrhocoridae (12) | ||||
| Pyrrhocoridae (1) | ND4L-P-T-T-T-T-T-T-ND6 | / | gene duplication, gene translocation | |
| Urostylididae (1) | CR-I-Q-M-ND2 | / | gene loss |
| Family | I-Q-M (Block A) | W-C-Y (Block A) | A-R-N-S-E-F (Block B) | T-P (Block C) | ND6-CYTB-S-ND1 (Block C) | rrnL-V-rrnS (Block C) | Whole Mitogenome |
|---|---|---|---|---|---|---|---|
| Aenictopecheidae | 0 | 0 | 0 | 25.0% | 50.0% | 0 | 5.0% |
| Enicocephalidae | 0 | 0 | 20.0% | 25.0% | 50.0% | 0 | 10.0% |
| Ceratocombidae | 0 | 0 | 20.0% | 0 | 0 | 50.0% | 5.0% |
| Gerridae | 0 | 0 | 20.0% | 0 | 0 | 0.0% | 5.0% |
| Nabidae | 28.6% | 0 | 0 | 0 | 0 | 0 | 10.0% |
| Pachynomidae | 0 | 20.0% | 20.0% | 0 | 50.0% | 0 | 5.0% |
| Reduviidae | 42.9% | 40.0% | 40.0% | 50.0% | 0 | 50.0% | 40.0% |
| Aradidae | 28.6% | 40.0% | 0 | 0 | 0 | 0 | 15.0% |
| Largidae | 0 | 0 | 0 | 25.0% | 0 | 0 | 5.0% |
| Pyrrhocoridae | 0 | 0 | 0 | 50.0% | 0 | 0 | 10.0% |
| Urostylididae | 14.3% | 0 | 0 | 0 | 0 | 0 | 5.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, F.; Li, H.; Xie, Q. Mitochondrial Genomes from Two Specialized Subfamilies of Reduviidae (Insecta: Hemiptera) Reveal Novel Gene Rearrangements of True Bugs. Genes 2021, 12, 1134. https://doi.org/10.3390/genes12081134
Ye F, Li H, Xie Q. Mitochondrial Genomes from Two Specialized Subfamilies of Reduviidae (Insecta: Hemiptera) Reveal Novel Gene Rearrangements of True Bugs. Genes. 2021; 12(8):1134. https://doi.org/10.3390/genes12081134
Chicago/Turabian StyleYe, Fei, Hu Li, and Qiang Xie. 2021. "Mitochondrial Genomes from Two Specialized Subfamilies of Reduviidae (Insecta: Hemiptera) Reveal Novel Gene Rearrangements of True Bugs" Genes 12, no. 8: 1134. https://doi.org/10.3390/genes12081134
APA StyleYe, F., Li, H., & Xie, Q. (2021). Mitochondrial Genomes from Two Specialized Subfamilies of Reduviidae (Insecta: Hemiptera) Reveal Novel Gene Rearrangements of True Bugs. Genes, 12(8), 1134. https://doi.org/10.3390/genes12081134