
Revealing the Viral Community in the Hadal Sediment of the New Britain Trench


 ,
, 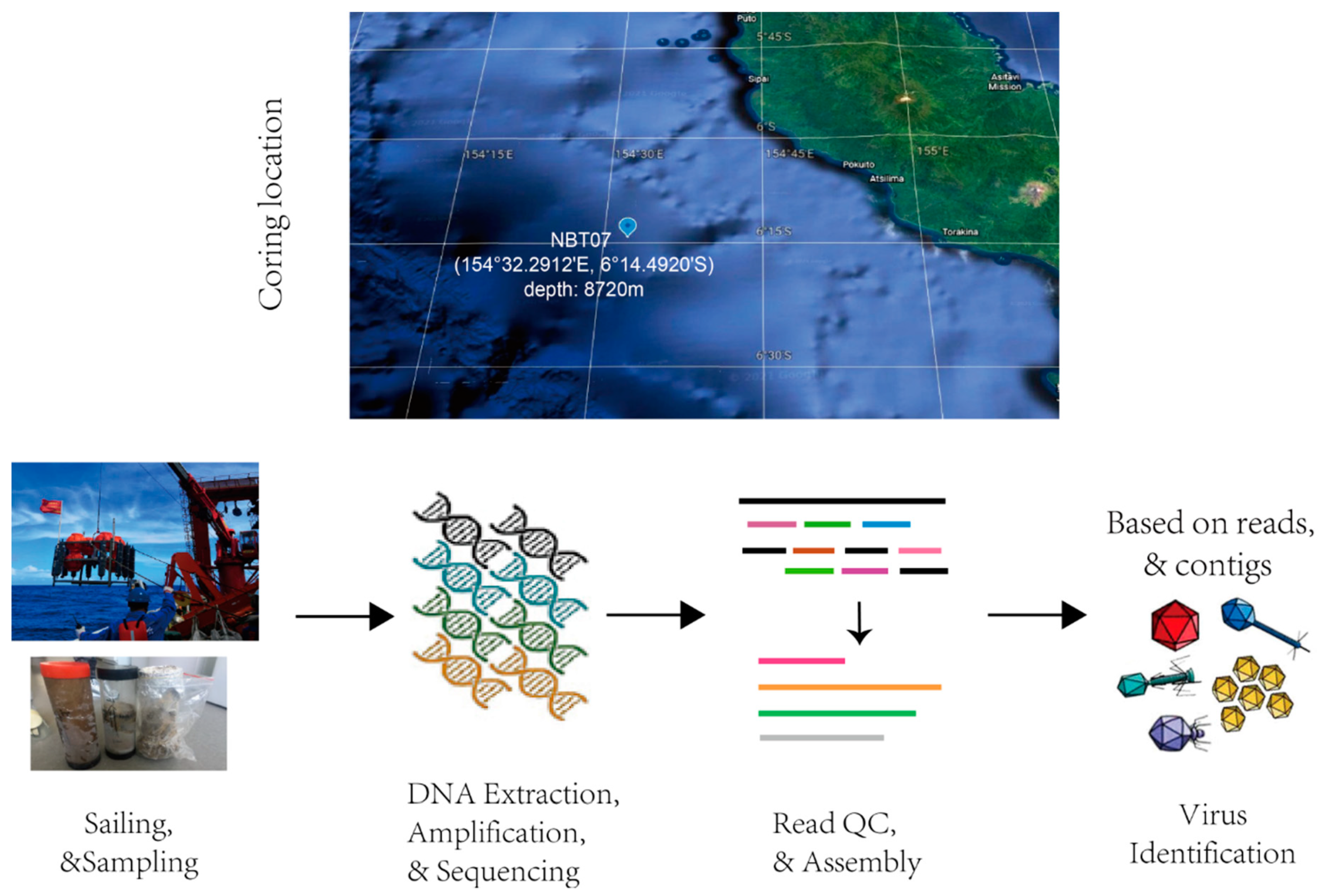{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Sampling Site Descriptions and Sample Collection
2.2. DNA Extraction, Library Construction, and Sequencing
2.3. Sequence Processing and Assembly
2.4. Taxonomic Classification of Reads and Determination of Relative Sequence Abundance
2.5. Recovering and Annotating Viral Contigs
2.6. Comparisons to Viral Sequences from Other Environments and Data Sets
2.7. Functional Viromics Analysis
3. Results and Discussion
3.1. Deep Sequencing of the Hadal Microbiome in the NBT Sediments and Identification of Viral Genome/Metavirome
3.2. Compositions of the Hadal Viruses in the NBT Sediment Communities
3.3. Comparative Analysis of Viromic Compositions between the NBT Sediment and Hadal Aquatic Habitats
3.4. Comparative Analysis of Viromic Compositions between the NBT Sediment and Bathypelagic and Terrestrial Habitats
3.5. Phylogenetic Diversity of the Hadal Virome in the NBT Sediment
3.6. Functional Analysis of the Hadal Viromics in NBT Sediments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamieson, A.J.; Fujii, T.; Mayor, D.J.; Solan, M.; De Prie, I.G. Hadal trenches: The ecology of the deepest places on Earth. Trends Ecol. Evol. 2010, 25, 190–197. [Google Scholar] [CrossRef]
- Peoples, L.M.; Sierra, D.; Oladayo, O.; Xia, Q.; Alex, N.; Jessica, B.; Allen, E.E.; Church, M.J.; Bartlett, D.H.; Hauke, S. Vertically distinct microbial communities in the Mariana and Kermadec trenches. PLoS ONE 2018, 13, e0195102. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, Y.; Lin, H.; Wang, X.; Li, M.; Liu, Y.; Yu, M.; Zhao, M.; Pedentchouk, N.; Lea-Smith, D.J.; et al. Proliferation of hydrocarbon-degrading microbes at the bottom of the Mariana Trench. Microbiome 2019, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Corinaldesi, C. New perspectives in benthic deep-sea microbial ecology. Front. Mar. Sci. 2015, 2, 17. [Google Scholar] [CrossRef]
- Danovaro, R.; Corinaldesi, C.; Rastelli, E.; Dell’Anno, A. Towards a better quantitative assessment of the relevance of deep-sea viruses, Bacteria and Archaea in the functioning of the ocean seafloor. Aquat. Microb. Ecol. 2015, 75, 81–90. [Google Scholar] [CrossRef]
- Suttle, C. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Winter, C.; Garcia, J.; Weinbauer, M.G.; Dubow, M.S.; Herndl, G.J. Comparison of Deep-Water Viromes from the Atlantic Ocean and the Mediterranean Sea. PLoS ONE 2014, 9, e100600. [Google Scholar] [CrossRef]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef]
- Danovaro, R.; Dell’Anno, A.; Corinaldesi, C.; Magagnini, M.; Noble, R.; Tamburini, C.; Weinbauer, M. Major viral impact on the functioning of benthic deep-sea ecosystems. Nature 2008, 454, 1084–1087. [Google Scholar] [CrossRef]
- Manea, E.; Dell’Anno, A.; Rastelli, E.; Tangherlini, M.; Corinaldesi, C. Viral infections boost prokaryotic biomass production and organic C cycling in hadal trench sediments. Front. Microbiol. 2019, 10, 1952. [Google Scholar] [CrossRef]
- Gallo, N.D.; James, C.; Kevin, H.; Patricia, F.; Douglas, H.B.; Lisa, A.L. Submersible- and lander-observed community patterns in the Mariana and New Britain trenches: Influence of productivity and depth on epibenthic and scavenging communities. Deep Sea Res. Part I Oceanogr. Res. Pap. 2015, 99, 119–133. [Google Scholar] [CrossRef]
- Davies, H.L.; Keene, J.B.; Hashimoto, K.; Joshima, M.; Stuart, J.E.; Tiffin, D.L. Bathymetry and canyons of the western Solomon Sea. Geo Mar. Lett. 1986, 6, 181–191. [Google Scholar] [CrossRef]
- Liu, R.; Wang, L.; Liu, Q.; Wang, Z.; Li, Z.; Fang, J.; Zhang, L.; Luo, M. Depth-resolved distribution of Particle-Attached and Free-Living bacterial communities in the water column of the New Britain Trench. Front. Microbiol. 2018, 9, 625. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Z.; Wang, L.; Li, Z.; Fang, J.; Wei, X.; Wei, W.; Cao, J.; Wei, Y.; Xie, Z. Bulk and active sediment prokaryotic communities in the Mariana and Mussau Trenches. Front. Microbiol. 2020, 11, 1521. [Google Scholar] [CrossRef]
- Wang, L.; Liu, R.; Wei, X.; Wang, Z.; Shen, Z.; Cao, J.; Wei, Y.; Xie, Z.; Chen, L.; Fang, J. Transitions in microbial communities along two sediment cores collected from the landward walls of the New Britain trench. Mar. Biol. 2020, 167, 172. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, J.; Li, J.; Zhang, L.; Xie, B.-B.; Chen, X.-L.; Zhang, Y.-Z. Depth-resolved variations of cultivable bacteria and their extracellular enzymes in the water column of the New Britain Trench. Front. Microbiol. 2018, 9, 135. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhou, H.; Huang, Y.; Xie, Z.; Zhang, M.; Wei, Y.; Li, J.; Ma, Y.; Luo, M.; Ding, W.; et al. Revealing the full biosphere structure and versatile metabolic functions in the deepest ocean sediment of the Challenger Deep. bioRxiv 2021. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.B.; Luo, X.; Qian, J.; Pang, X.H.; Song, J.Y. FastUniq: A Fast De Novo Duplicates Removal Tool for Paired Short Reads. PLoS ONE 2012, 7, e52249. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef]
- Paula, D.M.; Danczak, R.E.; Simon, R.; Jeroen, F.; Borton, M.A.; Wolfe, R.A.; Burris, M.N.; Wilkins, M.J. Viral and metabolic controls on high rates of microbial sulfur and carbon cycling in wetland ecosystems. Microbiome 2018, 6, 138. [Google Scholar]
- Emerson, J.B.; Simon, R.; Brum, J.R.; Benjamin, B.; Woodcroft, B.J.; Bin, J.H.; Singleton, C.M.; Solden, L.M.; Naas, A.; Boyd, J.A. Host-linked soil viral ecology along a permafrost thaw gradient. Nat. Microbiol. 2018, 3, 870–880. [Google Scholar] [CrossRef]
- Li, Z.; Pan, D.; Wei, G.; Pi, W.; Zhang, C.; Wang, J.-H.; Peng, Y.; Zhang, L.; Wang, Y.; Hubert, C.R.J.; et al. Deep sea sediments associated with cold seeps are a subsurface reservoir of viral diversity. ISME J. 2021. [Google Scholar] [CrossRef]
- Jang, H.B.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Yutin, N. Origin and evolution of eukaryotic large nucleo-cytoplasmic DNA viruses. Intervirology 2010, 53, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.S.; Rise, M.L.; Culley, A.I.; Steward, G.F. RNA viruses in the sea. FEMS Microbiol. Rev. 2009, 33, 295–323. [Google Scholar] [CrossRef]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.-M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef]
- Abrahão, J.; Silva, L.; Silva, L.S.; Khalil, J.Y.B.; Rodrigues, R.; Arantes, T.; Assis, F.; Boratto, P.; Andrade, M.; Kroon, E.G.; et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Filee, J.; Pouget, N.; Chandler, M. Phylogenetic evidence for extensive lateral acquisition of cellular genes by Nucleocytoplasmic large DNA viruses. BMC Evol. Biol. 2008, 8, 320. [Google Scholar] [CrossRef]
- Bekliz, M.; Colson, P.; La Scola, B. The Expanding Family of Virophages. Viruses 2016, 8, 317. [Google Scholar] [CrossRef]
- Fischer, M.G. The Virophage Family Lavidaviridae. Curr. Issues Mol. Biol. 2021, 40, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.M.; Hussain, F.A.; Yang, J.; Arevalo, P.; Brown, J.M.; Chang, W.K.; VanInsberghe, D.; Elsherbini, J.; Sharma, R.S.; Cutler, M.B.; et al. A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nature 2018, 554, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Labonté, J.; Suttle, C.A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013, 7, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, W.; Liu, Y.; Cai, M.; Luo, Z.; Li, M. Metagenomics Reveals Microbial Diversity and Metabolic Potentials of Seawater and Surface Sediment from a Hadal Biosphere at the Yap Trench. Front. Microbiol. 2018, 9, 2402. [Google Scholar] [CrossRef]
- Hiraoka, S.; Hirai, M.; Matsui, Y.; Makabe, A.; Minegishi, H.; Tsuda, M.; Juliarni Rastelli, E.; Danovaro, R.; Corinaldesi, C.; Kitahashi, T.; et al. Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments. ISME J. 2020, 14, 740–756. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Jin, M.; Guo, X.; Zhang, R.; Qu, W.; Gao, B.; Zeng, R. Diversities and potential biogeochemical impacts of mangrove soil viruses. Microbiome 2019, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Skennerton, C.T.; Angly, F.E.; Breitbart, M.; Bragg, L.; He, S.; McMahon, K.D.; Hugenholtz, P.; Tyson, G.W. Phage encoded H-NS: A potential achilles heel in the bacterial defence system. PLoS ONE 2011, 6, e20095. [Google Scholar] [CrossRef]
- Summer, E.J.; Enderle, C.J.; Ahern, S.J.; Gill, J.J.; Torres, C.P.; Appel, D.N.; Black, M.C.; Young, R.; Gonzalez, C.F. Genomic and Biological Analysis of Phage Xfas53 and Related Prophages of Xylella fastidiosa. J. Bacteriol. 2010, 192, 179. [Google Scholar] [CrossRef]
- Clark, A.J.; Pontes, M.; Jones, T.; Dale, C. A possible heterodimeric prophage-like element in the genome of the insect endosymbiont Sodalis glossinidius. J. Bacteriol. 2007, 189, 2949. [Google Scholar] [CrossRef][Green Version]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Chen, P.; Zhang, M.; Chen, J.; Fang, J.; Li, X. Revealing the Viral Community in the Hadal Sediment of the New Britain Trench. Genes 2021, 12, 990. https://doi.org/10.3390/genes12070990
Zhou H, Chen P, Zhang M, Chen J, Fang J, Li X. Revealing the Viral Community in the Hadal Sediment of the New Britain Trench. Genes. 2021; 12(7):990. https://doi.org/10.3390/genes12070990
Chicago/Turabian StyleZhou, Hui, Ping Chen, Mengjie Zhang, Jiawang Chen, Jiasong Fang, and Xuan Li. 2021. "Revealing the Viral Community in the Hadal Sediment of the New Britain Trench" Genes 12, no. 7: 990. https://doi.org/10.3390/genes12070990
APA StyleZhou, H., Chen, P., Zhang, M., Chen, J., Fang, J., & Li, X. (2021). Revealing the Viral Community in the Hadal Sediment of the New Britain Trench. Genes, 12(7), 990. https://doi.org/10.3390/genes12070990