
Genotype–Phenotype Correlations in Angelman Syndrome

Abstract
:1. Introduction
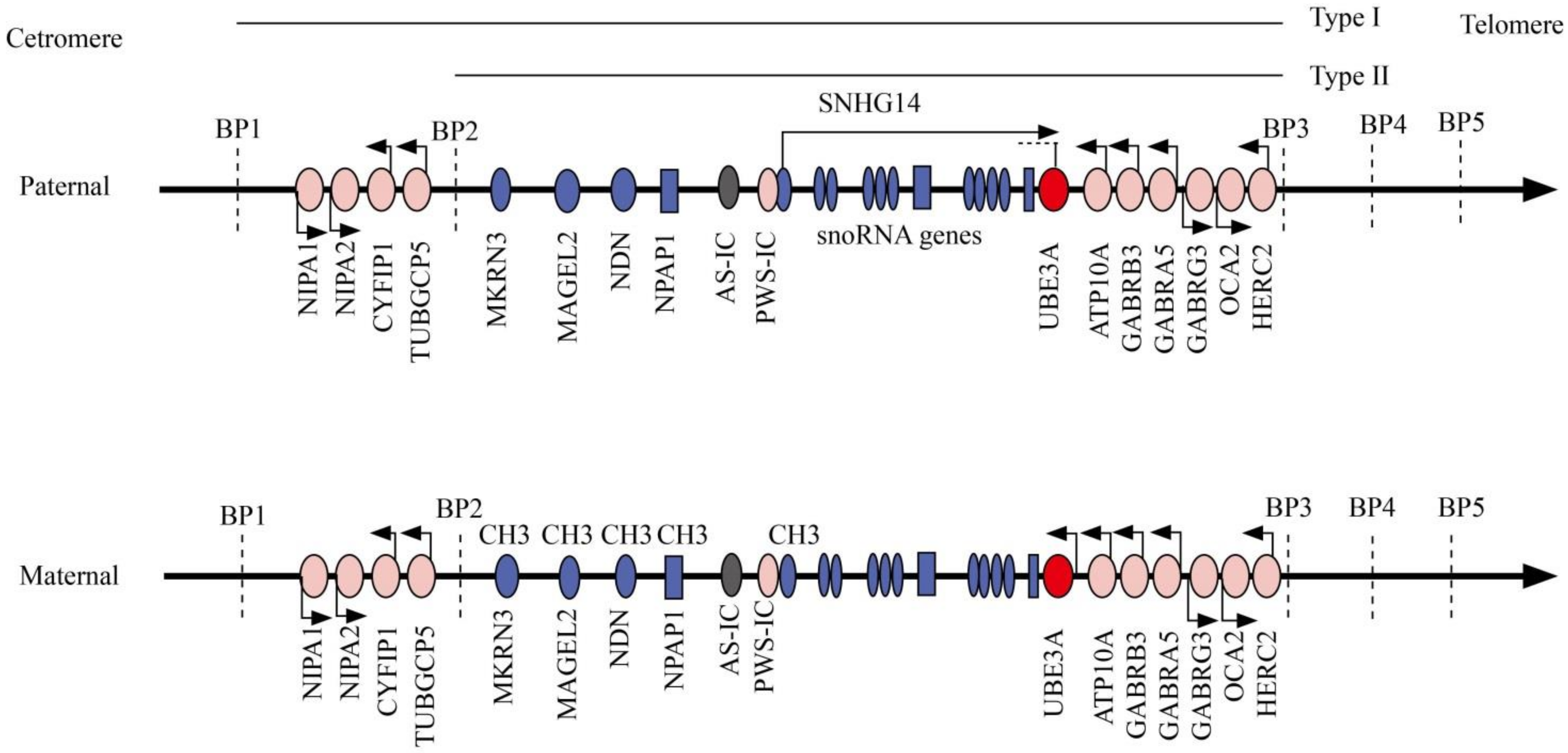
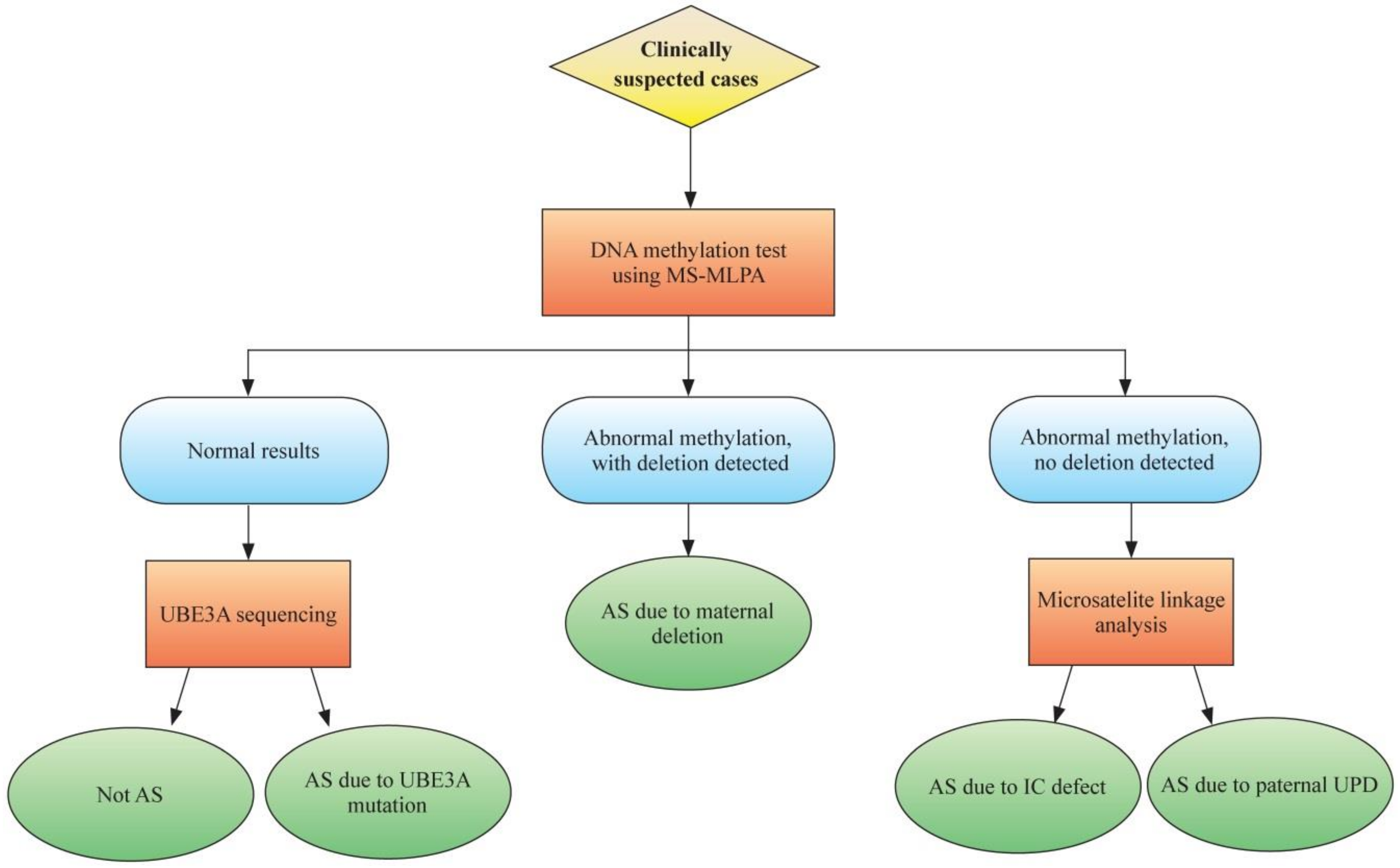
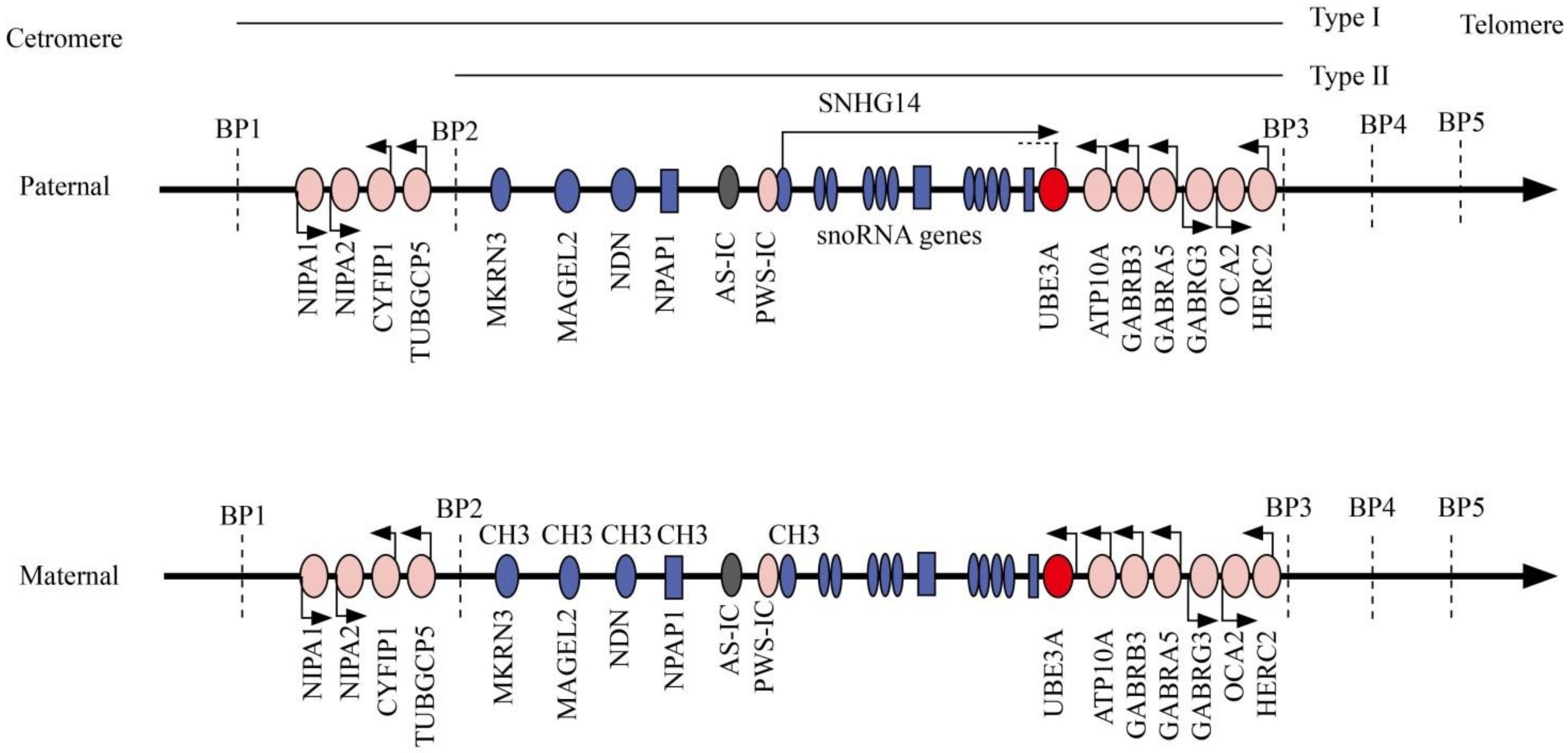
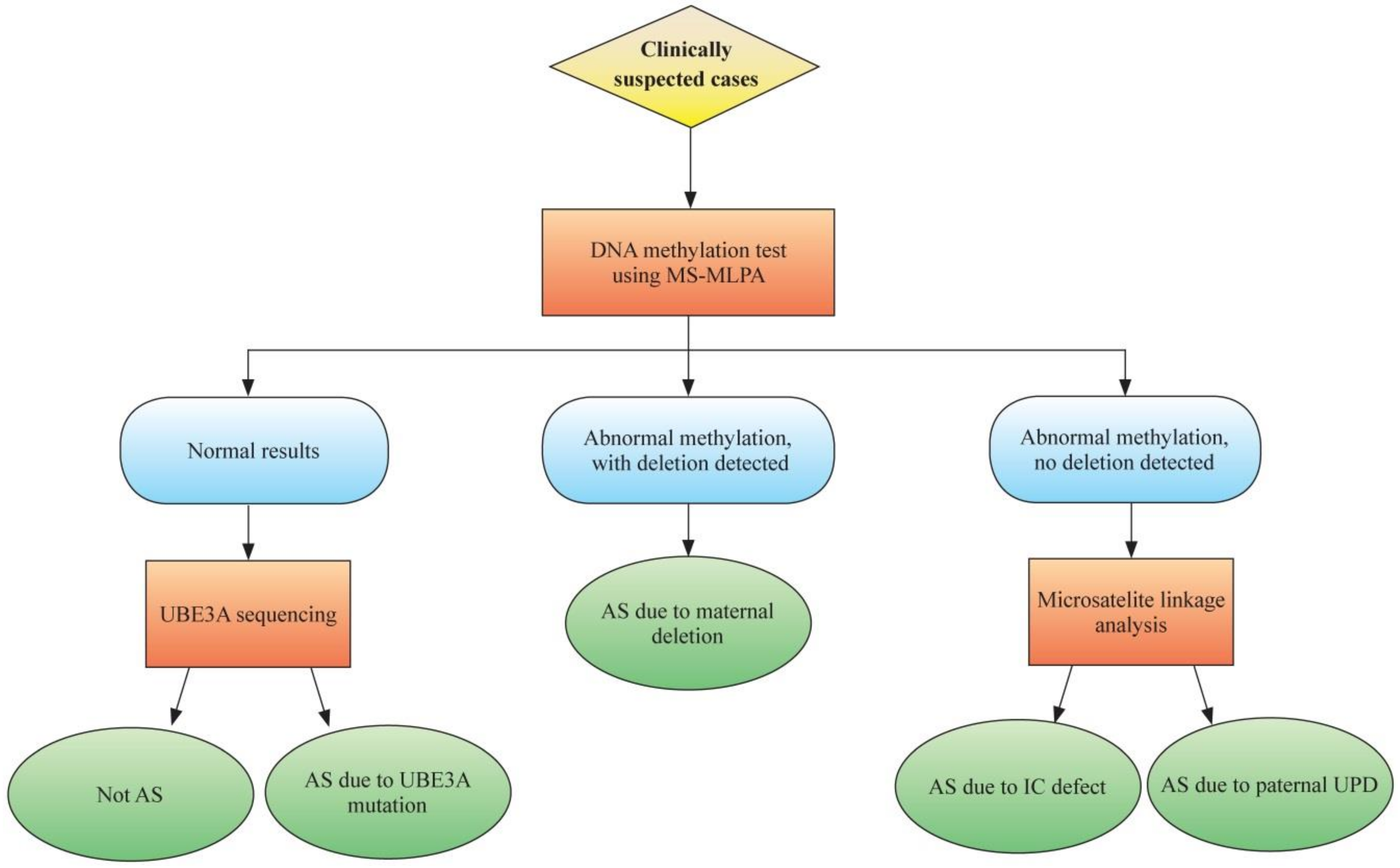
2. Molecular Genetics and Diagnostics
3. Genotype–Phenotype Correlation in Angelman Syndrome
3.1. AS Due to Maternal del15q11–13
3.2. AS Due to Paternal Uniparental Disomy for Chromosome15q11–q13
3.3. AS Due to Imprinting Defect
3.4. AS Due to Pathogenic UBE3A Mutation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, C.A.; Driscoll, D.J.; Dagli, A.I. Clinical and genetic aspects of Angelman syndrome. Genet. Med. 2010, 12, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Maranga, C.; Fernandes, T.G.; Bekman, E.; Da Rocha, S.T. Angelman syndrome: A journey through the brain. FEBS J. 2020, 287, 2154–2175. [Google Scholar] [CrossRef]
- Buonfiglio, D.; Hummer, D.L.; Armstrong, A.; Ehlen, J.C.; DeBruyne, J.P. Angelman syndrome and melatonin: What can they teach us about sleep regulation. J. Pineal Res. 2020, 69, 12697. [Google Scholar] [CrossRef]
- Angelman, H. “Puppet children”. A report of three cases. Dev. Med. Child. Neurol. 1965, 7, 688. [Google Scholar] [CrossRef]
- Williams, C.A. Neurological aspects of the Angelman syndrome. Brain Dev. 2005, 27, 88–94. [Google Scholar] [CrossRef]
- Thibert, R.L.; Larson, A.M.; Hsieh, D.T.; Raby, A.R.; Thiele, E.A. Neurologic Manifestations of Angelman Syndrome. Pediatr. Neurol. 2013, 48, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Bird, L. Angelman syndrome: Review of clinical and molecular aspects. Appl. Clin. Genet. 2014, 7, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Keute, M.; Miller, M.T.; Krishnan, M.L.; Sadhwani, A.; Chamberlain, S.; Thibert, R.L.; Tan, W.-H.; Bird, L.M.; Hipp, J.F. Angelman Syndrome Genotypes Manifest Varying Degrees of Clinical Severity and Developmental Impairment. Available online: https://www.nature.com/articles/s41380-020-0858-6 (accessed on 13 August 2020).
- Kaplan, L.C.; Wharton, R.; Elias, E.; Mandell, F.; Donlon, T.; Latt, S.A.; Opitz, J.M.; Reynolds, J.F. Clinical heterogeneity associated with deletions in the long arm of chromosome 15: Report of 3 new cases and their possible genetic significance. Am. J. Med. Genet. 1987, 28, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.D. Genomic imprinting and uniparental disomy in Angelman and Prader-Willi syndromes: A review. Am. J. Med. Genet. 1993, 46, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Saitoh, S.; Gross, S.; Dittrich, B.; Schwartz, S.; Nicholls, R.D.; Horsthemke, B. Inherited microdeletions in the Angelman and Prader–Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 1995, 9, 395–400. [Google Scholar] [CrossRef]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef]
- Peters, S.U.; Horowitz, L.; Barbieri-Welge, R.; Taylor, J.L.; Hundley, R.J. Longitudinal follow-up of autism spectrum features and sensory behaviors in Angelman syndrome by deletion class. J. Child Psychol. Psychiatry 2011, 53, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Gentile, J.K.; Tan, W.-H.; Horowitz, L.T.; Bacino, C.A.; Skinner, S.A.; Barbieri-Welge, R.; Bauer-Carlin, A.; Beaudet, A.L.; Bichell, T.J.; Lee, H.-S.; et al. A Neurodevelopmental Survey of Angelman Syndrome with Genotype-Phenotype Correlations. J. Dev. Behav. Pediatr. 2010, 31, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.-H.; Bacino, C.A.; Skinner, S.A.; Anselm, I.; Barbieri-Welge, R.; Bauer-Carlin, A.; Beaudet, A.L.; Bichell, T.J.; Gentile, J.K.; Glaze, D.G.; et al. Angelman syndrome: Mutations influence features in early childhood. Am. J. Med. Genet. Part A 2011, 155, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, K.D.; Varela, M.C.; Koiffmann, C.P.; Andrade, J.Q.; Grossmann, R.; Kok, F.; Marques-Dias, M.J. Angelman syndrome caused by deletion: A genotype–phenotype correlation determined by breakpoint. Epilepsy Res. 2013, 105, 234–239. [Google Scholar] [CrossRef]
- Margolis, S.S.; Sell, G.L.; Zbinden, M.A.; Bird, L.M. Angelman Syndrome. Neurotherapeutics 2015, 12, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Buiting, K.; Williams, C.; Horsthemke, K.B.B. Angelman syndrome—Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Andersen, W.H.; Rasmussen, R.K.; Strømme, P. Levels of cognitive and linguistic development in Angelman syndrome: A study of 20 children. Logoped. Phoniatr. Vocol. 2001, 26, 2–9. [Google Scholar] [CrossRef]
- Boyd, S.; Harden, A.; Patton, M.A. The EEG in early diagnosis of the Angelman (Happy Puppet) syndrome. Eur. J. Nucl. Med. Mol. Imaging 1988, 147, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Dan, B.; Boyd, S. Angelman Syndrome Reviewed from a Neurophysiological Perspective. The UBE3A-GABRB3 Hypothesis. Neuropediatrics 2003, 34, 169–176. [Google Scholar] [PubMed]
- Born, H.A.; Martinez, L.A.; Levine, A.T.; Harris, S.E.; Mehra, S.; Lee, W.L.; Dindot, S.V.; Nash, K.R.; Silverman, J.L.; Segal, D.J.; et al. Early Developmental EEG and Seizure Phenotypes in a Full Gene Deletion of Ubiquitin Protein Ligase E3A Rat Model of Angelman Syndrome. Eneuro 2021, 8, ENEURO.0345-20.2020. [Google Scholar] [CrossRef]
- Micheletti, S.; Palestra, F.; Martelli, P.; Accorsi, P.; Galli, J.; Giordano, L.; Trebeschi, V.; Fazzi, E. Neurodevelopmental profile in Angelman syndrome: More than low intelligence quotient. Ital. J. Pediatr. 2016, 42, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Dan, B.; Pelc, K.; Cheron, G. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr. Dis. Treat. 2008, 4, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Bruni, O.; Ferri, R.; D’Agostino, G.; Miano, S.; Roccella, M.; Elia, M. Sleep disturbances in Angelman syndrome: A questionnaire study. Brain Dev. 2004, 26, 233–240. [Google Scholar] [CrossRef]
- Didden, R.; Korzilius, H.; Smits, M.G.; Curfs, L.M.G. Sleep Problems in Individuals with Angelman Syndrome. Am. J. Ment. Retard. 2004, 109, 275–284. [Google Scholar] [CrossRef]
- Walz, N.C.; Beebe, D.; Byars, K. Sleep in Individuals with Angelman Syndrome: Parent Perceptions of Patterns and Problems. Am. J. Ment. Retard. 2005, 110, 243–252. [Google Scholar] [CrossRef]
- Pelc, K.; Cheron, G.; Boyd, S.; Dan, B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008, 9, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Takaesu, Y.; Komada, Y.; Inoue, Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients. Sleep Med. 2012, 13, 1164–1170. [Google Scholar] [CrossRef]
- Williams, C.A.; Beaudet, A.L.; Clayton-Smith, J.; Knoll, J.H.; Kyllerman, M.; Laan, L.A.; Magenis, R.E.; Moncla, A.; Schinzel, A.A.; Summers, J.A.; et al. Angelman syndrome 2005: Updated consensus for diagnostic criteria. Am. J. Med. Genet. Part A 2006, 140A, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Nakao, M.; Sutcliffe, J.S.; Durtschl, B.; Mutlrangura, A.; Ledbetter, D.H.; Beaudet, A.L. Imprinting analysis of three genes in the Prader—Willi/Angelman region: SNRPN, E6-associated protein, and PAR-2 (D15S225E). Hum. Mol. Genet. 1994, 3, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.Y.; Futtner, C.R.; Chamberlain, S.J.; Johnstone, K.A.; Resnick, J.L. Transcription Is Required to Establish Maternal Imprinting at the Prader-Willi Syndrome and Angelman Syndrome Locus. PLoS Genet. 2011, 7, e1002422. [Google Scholar] [CrossRef] [Green Version]
- Beygo, J.; Buiting, K.; Ramsden, S.C.; Ellis, R.; Clayton-Smith, J.; Kanber, D. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur. J. Hum. Genet. 2019, 27, 1326–1340. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.A.; Ferreira, I.R.; Cintra, H.A.; Gomes, L.H.F.; Guida, L.D.C. Genotype-Phenotype Relationships and Endocrine Findings in Prader-Willi Syndrome. Front. Endocrinol. 2019, 10, 864. [Google Scholar] [CrossRef] [Green Version]
- Glenn, C.C.; Saitoh, S.; Jong, M.T.; Filbrandt, M.M.; Surti, U.; Driscoll, D.J.; Nicholls, R.D. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am. J. Hum. Genet. 1996, 58, 335–346. [Google Scholar]
- Georgieva, B.; Atemin, S.; Todorova, A.; Todorov, T.; Miteva, A.; Avdjieva-Tzavella, D.; Mitev, V. Molecular-Genetic Diagnostics of Angelman Syndrome—The Bulgarian Experience. Acta Med. Bulg. 2020, 47, 9–16. [Google Scholar] [CrossRef]
- Judson, M.C.; Sosa-Pagan, J.O.; Del Cid, W.A.; Han, J.E.; Philpot, B.D. Allelic specificity of Ube3a Expression In The Mouse Brain During Postnatal Development. J. Comp. Neurol. 2014, 522, 1874–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, S.J.; Chen, P.-F.; Ng, K.Y.; Bourgois-Rocha, F.; Lemtiri-Chlieh, F.; Levine, E.; Lalande, M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader–Willi syndromes. Proc. Natl. Acad. Sci. USA 2010, 107, 17668–17673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaSalle, J.M.; Reiter, L.T.; Chamberlain, S.J. Epigenetic regulation ofUBE3Aand roles in human neurodevelopmental disorders. Epigenomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Heussler, H.S. Emerging Therapies and challenges for individuals with Angelman syndrome. Curr. Opin. Psychiatry 2021, 34, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zaaroor-Regev, D.; de Bie, P.; Scheffner, M.; Noy, T.; Shemer, R.; Heled, M.; Stein, I.; Pikarsky, E.; Ciechanover, A. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6788–6793. [Google Scholar] [CrossRef] [Green Version]
- Daily, J.; Smith, A.G.; Weeber, E.J. Spatial and temporal silencing of the human maternal UBE3A gene. Eur. J. Paediatr. Neurol. 2012, 16, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Mulherkar, S.A.; Jana, N.R. Loss of dopaminergic neurons and resulting behavioural deficits in mouse model of Angelman syndrome. Neurobiol. Dis. 2010, 40, 586–592. [Google Scholar] [CrossRef]
- Moreira-De-Sá, A.; Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Tomé Ângelo, R.; Cunha, R.A.; Canas, P.M. Motor Deficits Coupled to Cerebellar and Striatal Alterations in Ube3am−/p+ Mice Modelling Angelman Syndrome Are Attenuated by Adenosine A2A Receptor Blockade. Mol. Neurobiol. 2021, 58, 2543–2557. [Google Scholar] [CrossRef]
- Moreira-De-Sá, A.; Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Tomé Ângelo, R.; Cunha, R.A.; Canas, P.M. Adenosine A2A receptors format long-term depression and memory strategies in a mouse model of Angelman syndrome. Neurobiol. Dis. 2020, 146, 105137. [Google Scholar] [CrossRef] [PubMed]
- Cheron, G.; Mã¡rquez-Ruiz, J.; Kishino, T.; Dan, B. Disruption of the LTD dialogue between the cerebellum and the cortex in Angelman syndrome model: A timing hypothesis. Front. Syst. Neurosci. 2014, 8, 221. [Google Scholar] [CrossRef] [Green Version]
- Madaan, M.; Mendez, M.D. Angelman Syndrome; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Mahmoud, R.; Singh, P.; Weiss, L.; Lakatos, A.; Oakes, M.; Hossain, W.; Butler, M.G.; Kimonis, V. Newborn screening for Prader–Willi syndrome is feasible: Early diagnosis for better outcomes. Am. J. Med. Genet. Part A 2018, 179, 29–36. [Google Scholar] [CrossRef]
- Ferreira, I.R.; Costa, R.A.; Gomes, L.H.F.; Cunha, W.D.D.S.; Tyszler, L.S.; Freitas, S.; Junior, J.C.L.; De Vasconcelos, Z.F.M.; Nicholls, R.D.; Guida, L.D.C. A newborn screening pilot study using methylation-sensitive high resolution melting on dried blood spots to detect Prader-Willi and Angelman syndromes. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stalker, H.J.; Williams, C.A. Genetic counseling in Angelman syndrome: The challenges of multiple causes. Am. J. Med. Genet. 1998, 77, 54–59. [Google Scholar] [CrossRef]
- Sonzogni, M.; Hakonen, J.; Kleijn, M.B.; Silva-Santos, S.; Judson, M.C.; Philpot, B.D.; Van Woerden, G.M.; Elgersma, Y. Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol. Autism 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Elgersma, Y.; Sonzogni, M. UBE3A reinstatement as a disease-modifying therapy for Angelman syndrome. Dev. Med. Child Neurol. 2021, 63, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Lossie, A.C.; Whitney, M.M.; Amidon, D.; Dong, H.J.; Chen, P.; Theriaque, D.; Hutson, A.; Nicholls, R.D.; Zori, R.T.; Williams, C.A.; et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 2001, 38, 834–845. [Google Scholar] [CrossRef] [Green Version]
- Sadhwani, A.; Wheeler, A.; Gwaltney, A.; Peters, S.U.; Barbieri-Welge, R.L.; Horowitz, L.T.; Noll, L.M.; Hundley, R.J.; Bird, L.M.; Tan, W.-H. Developmental Skills of Individuals with Angelman Syndrome Assessed Using the Bayley-III. Available online: http://link.springer.com/article/10.1007/s10803-020-04861-1 (accessed on 30 January 2021).
- Sahoo, T.; Peters, S.U.; Madduri, N.S.; Glaze, D.G.; German, J.R.; Bird, L.M.; Barbieri-Welge, R.; Bichell, T.J.; Beaudet, A.L.; Bacino, C.A. Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: Genotype-phenotype correlations. J. Med. Genet. 2006, 43, 512–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, T.; Bacino, C.A.; German, J.R.; Shaw, C.; Bird, L.M.; Kimonis, V.; Anselm, I.; Waisbren, S.; Beaudet, A.L.; Peters, S.U. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: Molecular characterization and genotype–phenotype correlations. Eur. J. Hum. Genet. 2007, 15, 943–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela, M.C.; Kok, F.; Otto, P.A.; Koiffmann, C.P. Phenotypic variability in Angelman syndrome: Comparison among different deletion classes and between deletion and UPD subjects. Eur. J. Hum. Genet. 2004, 12, 987–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, L.G.B.; Thaulov, P.; Trillingsgaard, A.; Christensen, R.; Vogel, I.; Hertz, J.M.; Østergaard, J.R. Neurodevelopmental outcome in Angelman syndrome: Genotype–phenotype correlations. Res. Dev. Disabil. 2014, 35, 1742–1747. [Google Scholar] [CrossRef] [PubMed]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Qual. Life Res. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Roche, L.; Heussler, H. Parent perceptions, beliefs, and fears around genetic treatments and cures for children with Angelman syndrome. Am. J. Med. Genet. Part A 2020, 182, 1716–1724. [Google Scholar] [CrossRef]
- Moncla, A.; Malzac, P.; Livet, M.; Voelckel, M.; Mancini, J.; Delaroziere, J.C.; Philip, N.; Mattei, J.F. Angelman syndrome resulting from UBE3A mutations in 14 patients from eight families: Clinical manifestations and genetic counselling. J. Med. Genet. 1999, 36, 554–560. [Google Scholar]
- Peters, S.U.; Goddard-Finegold, J.; Beaudet, A.L.; Madduri, N.; Turcich, M.; Bacino, C.A. Cognitive and adaptive behavior profiles of children with Angelman syndrome. Am. J. Med. Genet. 2004, 128A, 110–113. [Google Scholar] [CrossRef]
- Moncla, A.; Malzac, P.; Voelckel, M.-A.; Auquier, P.; Girardot, L.; Mattei, M.-G.; Philip, N.; Mattei, J.-F.; Lalande, M.; Livet, M.-O. Phenotype–genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur. J. Hum. Genet. 1999, 7, 131–139. [Google Scholar] [CrossRef]
- Luk, H.; Lo, I.F. Angelman syndrome in Hong Kong Chinese: A 20 years’ experience. Eur. J. Med. Genet. 2016, 59, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Shaaya, E.A.; Grocott, O.R.; Laing, O.; Thibert, R.L. Seizure treatment in Angelman syndrome: A case series from the Angelman Syndrome Clinic at Massachusetts General Hospital. Epilepsy Behav. 2016, 60, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D. Epilepsy in Angelman syndrome: A scoping review. Brain Dev. 2021, 43, 32–44. [Google Scholar] [CrossRef]
- Heald, M.; Adams, D.; Walls, E.; Oliver, C. Refining the Behavioral Phenotype of Angelman Syndrome: Examining Differences in Motivation for Social Contact Between Genetic Subgroups. Front. Behav. Neurosci. 2021, 15, 618271. [Google Scholar] [CrossRef]
- Vendrame, M.; Loddenkemper, T.; Zarowski, M.; Gregas, M.; Shuhaiber, H.; Sarco, D.P.; Morales, A.; Nespeca, M.; Sharpe, C.; Haas, K.; et al. Analysis of EEG patterns and genotypes in patients with Angelman syndrome. Epilepsy Behav. 2012, 23, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Mertz, L.G.; Christensen, R.; Vogel, I.; Hertz, J.M.; Østergaard, J.R. Eating behavior, prenatal and postnatal growth in Angelman syndrome. Res. Dev. Disabil. 2014, 35, 2681–2690. [Google Scholar] [CrossRef]
- Brennan, M.-L.; Adam, M.P.; Seaver, L.H.; Myers, A.; Schelley, S.; Zadeh, N.; Hudgins, L.; Bernstein, J.A. Increased body mass in infancy and early toddlerhood in Angelman syndrome patients with uniparental disomy and imprinting center defects. Am. J. Med. Genet. Part A 2015, 167, 142–146. [Google Scholar] [CrossRef]
- Horsthemke, B.; Buiting, K. Imprinting defects on human chromosome 15. Cytogenet. Genome Res. 2006, 113, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Saito, Y.; Honda, R.; Saito, T.; Sugai, K.; Matsuda, Y.; Miyatake, C.; Takeshita, E.; Ishiyama, A.; Komaki, H.; et al. Episodic tremors representing cortical myoclonus are characteristic in Angelman syndrome due to UBE3A mutations. Brain Dev. 2015, 37, 216–222. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Consistent (100%) | Frequent (>80%) | Associated (<80%) |
|---|---|---|
|
|
|
| Major Aspects | AS Due to Maternal del15q11–13 | AS Due to Non-Deletion | ||
|---|---|---|---|---|
| Paternal Uniparental Disomy for Chromosome15q11–q13 (UPD) | Imprinting Defect | Pathogenic UBE3A Mutation | ||
| Development | More delayed across all development domains than other types Cognitive skills lower than other types Delayed gross and fine motor skills more severe Reduced developmental age regarding visual perception, receptive language, and expressive language | Higher overall age equivalent scores and growth score equivalents than deleion type but lower than UBE3A mutation subtype; Better development and expressive language ability in patients with UPD and imprinting defect Higher scores and greater rates of skill attainment in all development domains in patients UBE3A mutation | ||
| Seizures | More common and severe in the deletion group | Lower prevalence of epilepsy, and more with late-onset seizures. UPD subtype has the lowest frequency of epilepsy and exhibits the least severe epilepsy phenotype The severity of epilepsy in the UBE3A mutation subtype ranks second after the deletion subtype | ||
| Behavior | Lower response rates to the social reinforcement paradigm than other types | The imprinting defect a high rate of reinforcement by social stimuli. Patients with UBE3A mutations | ||
| Sleep | Common in all subtypes but Sleep problems are more prevalent in children with UPD and UBE3A mutations | |||
| Others | Higher rate of hypopigmentation | UPD and imprinting defects have a higher risk of obesity than deletion type | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Shu, X.; Mao, S.; Wang, Y.; Du, X.; Zou, C. Genotype–Phenotype Correlations in Angelman Syndrome. Genes 2021, 12, 987. https://doi.org/10.3390/genes12070987
Yang L, Shu X, Mao S, Wang Y, Du X, Zou C. Genotype–Phenotype Correlations in Angelman Syndrome. Genes. 2021; 12(7):987. https://doi.org/10.3390/genes12070987
Chicago/Turabian StyleYang, Lili, Xiaoli Shu, Shujiong Mao, Yi Wang, Xiaonan Du, and Chaochun Zou. 2021. "Genotype–Phenotype Correlations in Angelman Syndrome" Genes 12, no. 7: 987. https://doi.org/10.3390/genes12070987
APA StyleYang, L., Shu, X., Mao, S., Wang, Y., Du, X., & Zou, C. (2021). Genotype–Phenotype Correlations in Angelman Syndrome. Genes, 12(7), 987. https://doi.org/10.3390/genes12070987