
Mitogenomes Reveal Two Major Influxes of Papuan Ancestry across Wallacea Following the Last Glacial Maximum and Austronesian Contact

 ,
,  , , ,
, , ,  ,
,  and
and 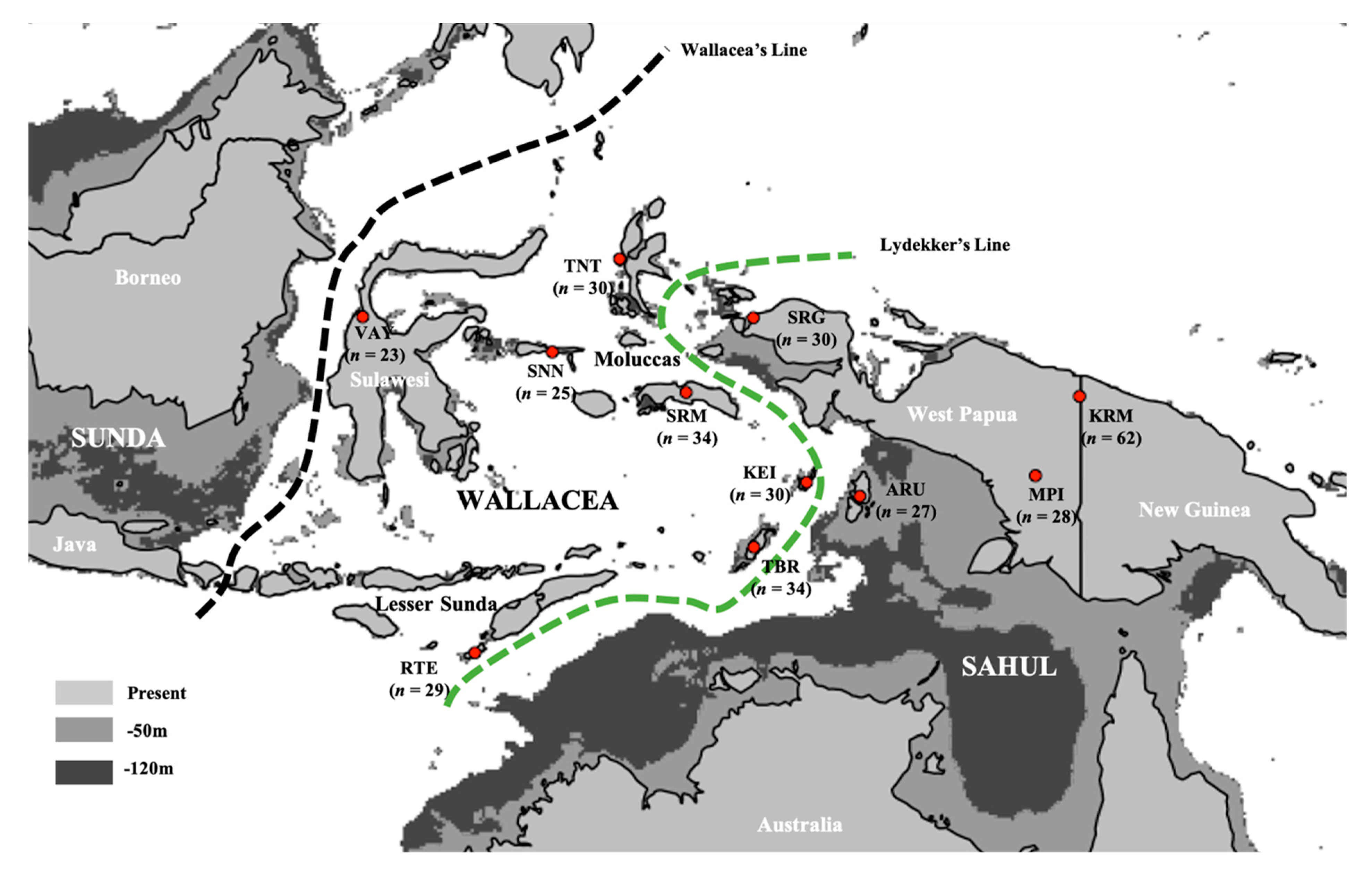{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Ethics
2.2. Mitochondrial Sequence Generation
2.3. Combined Wallacea–Sahul Dataset
2.4. Phylogenetic Parameter Estimation
2.5. Using Ancestral Node Dates from Geographically Exclusive Clades to Infer Demographic History
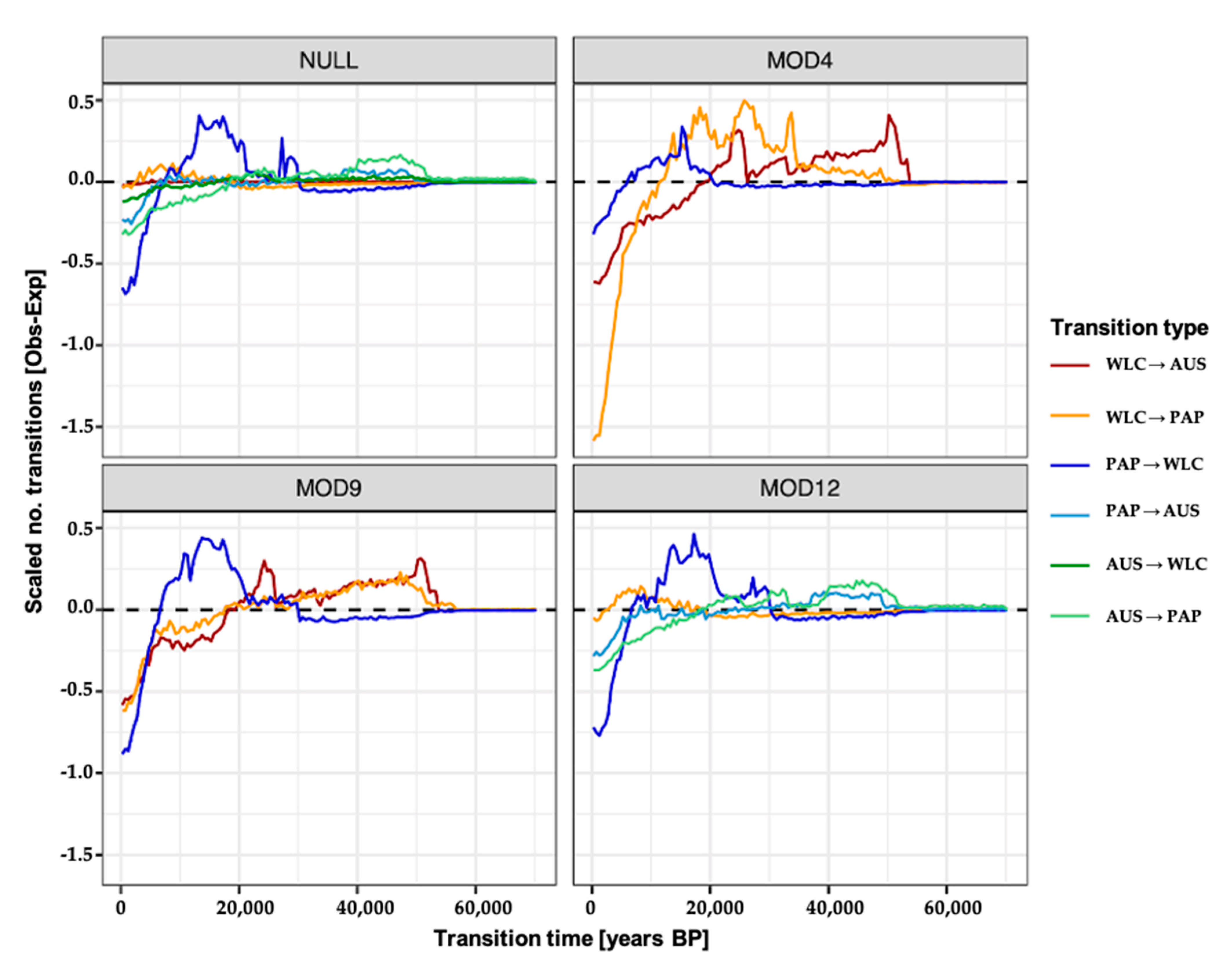
2.6. Migration Model Inference and Testing
2.7. Simulating and Estimating the Timing of Migration Events
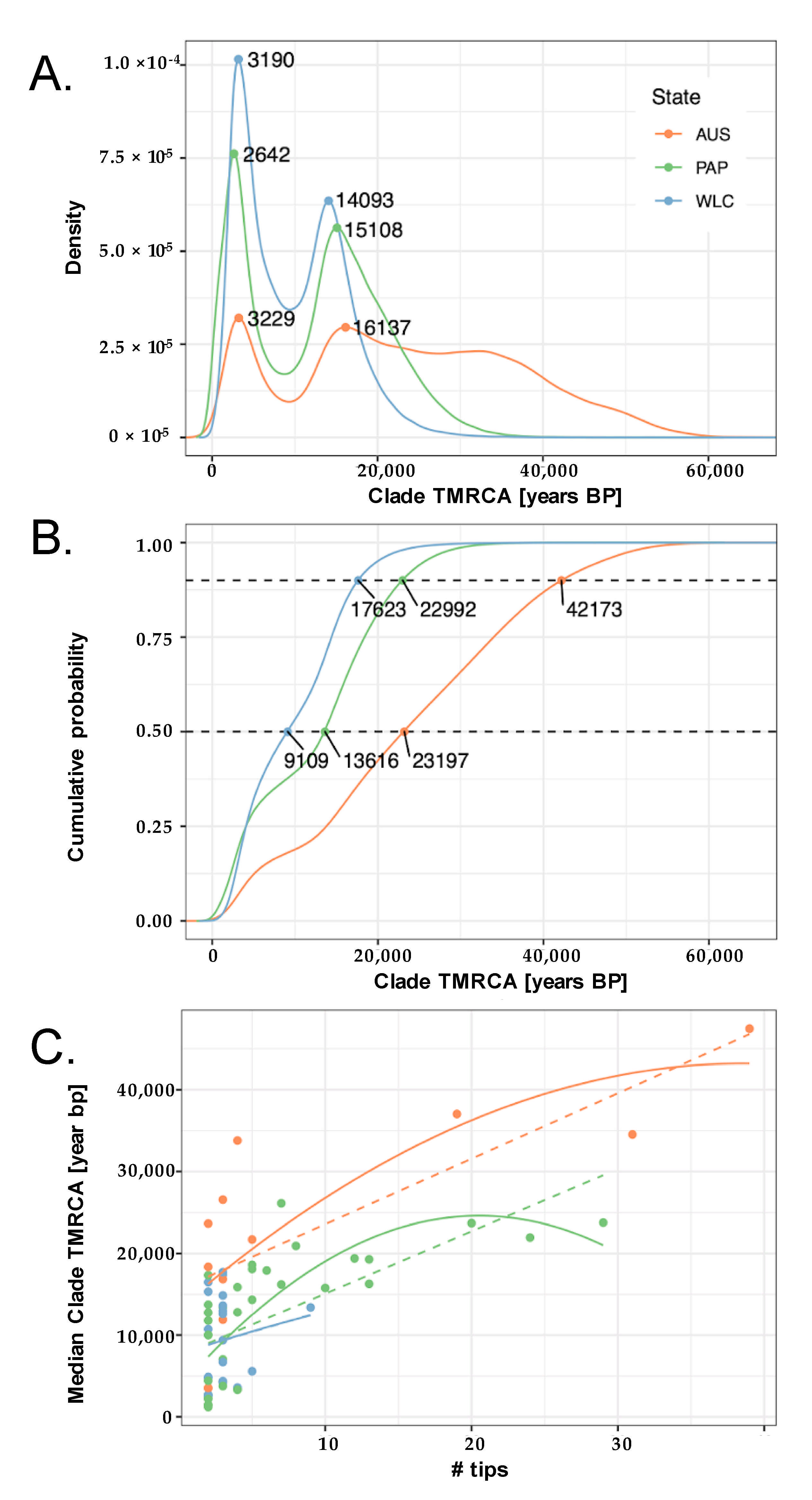
3. Results
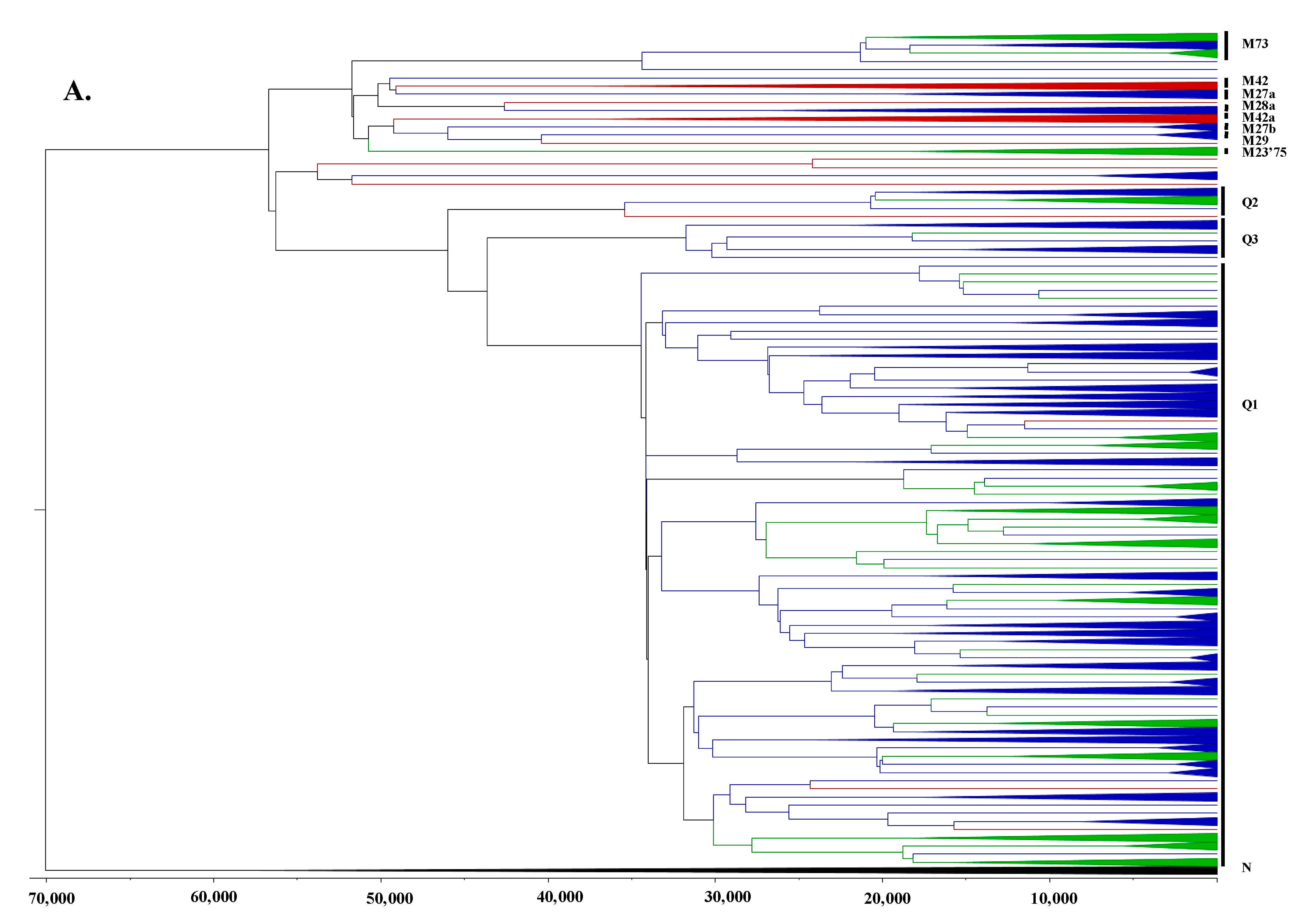
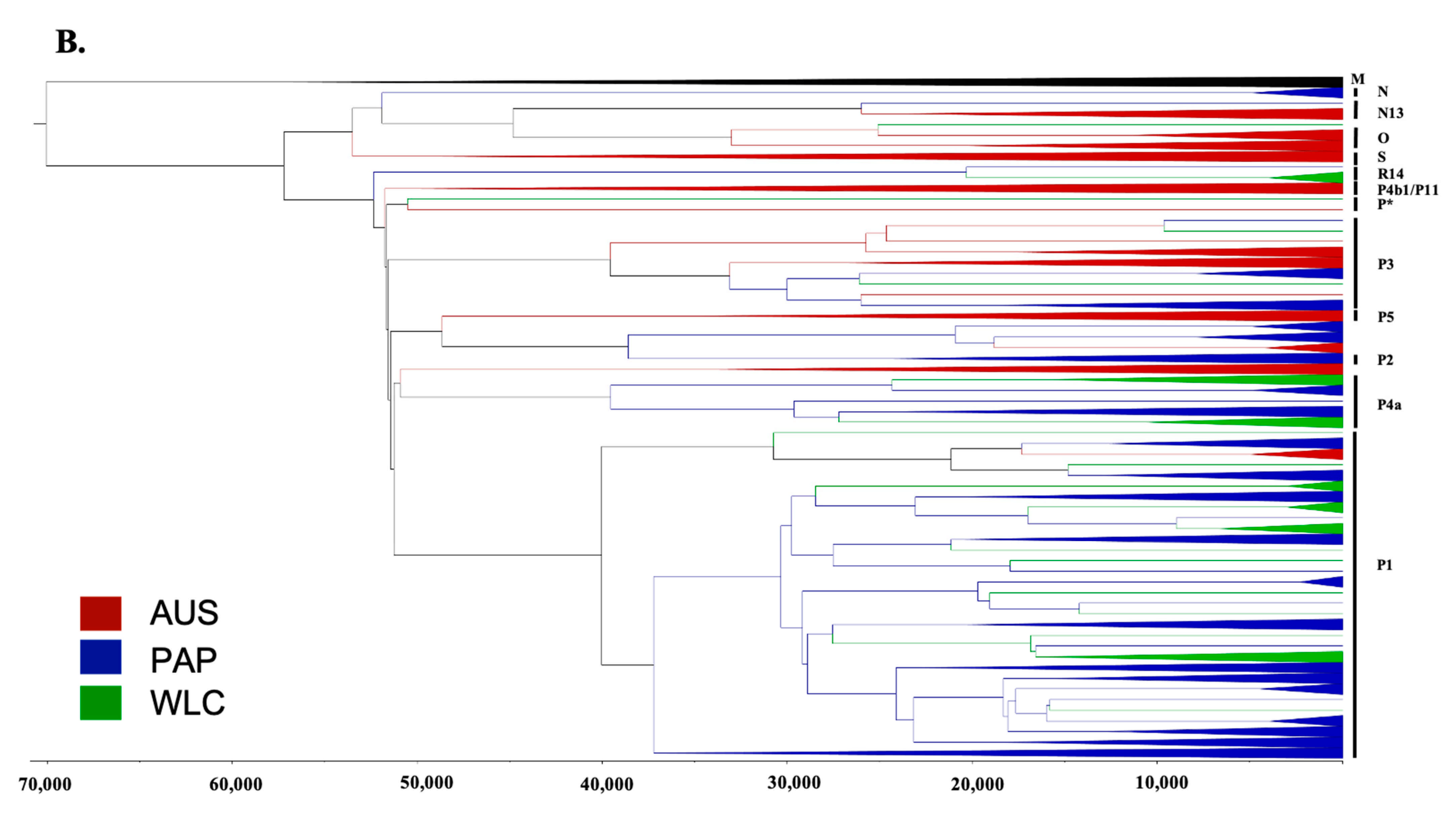
3.1. Summary of New Mitochondrial Haplogroups from Wallacea and West Papua
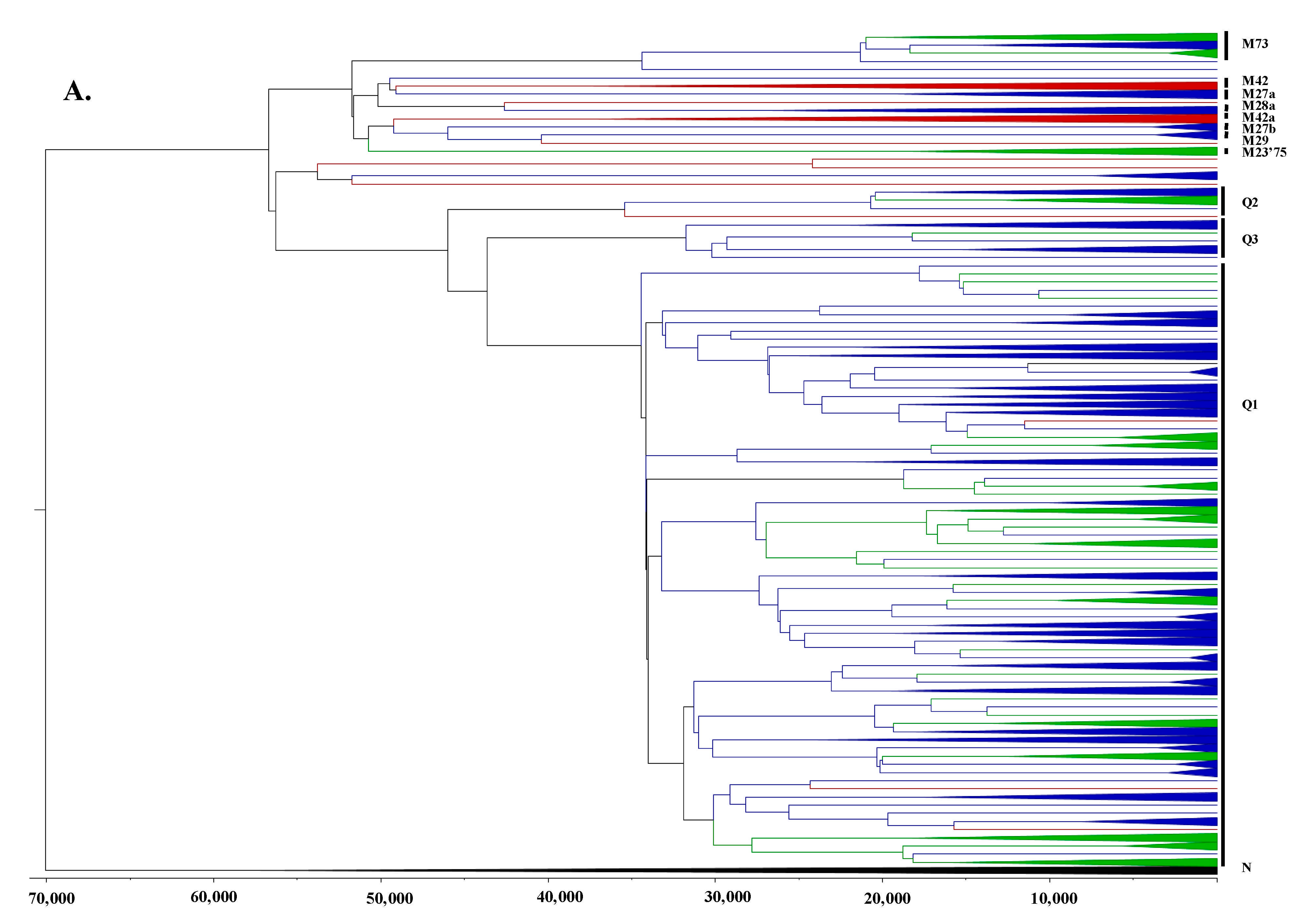
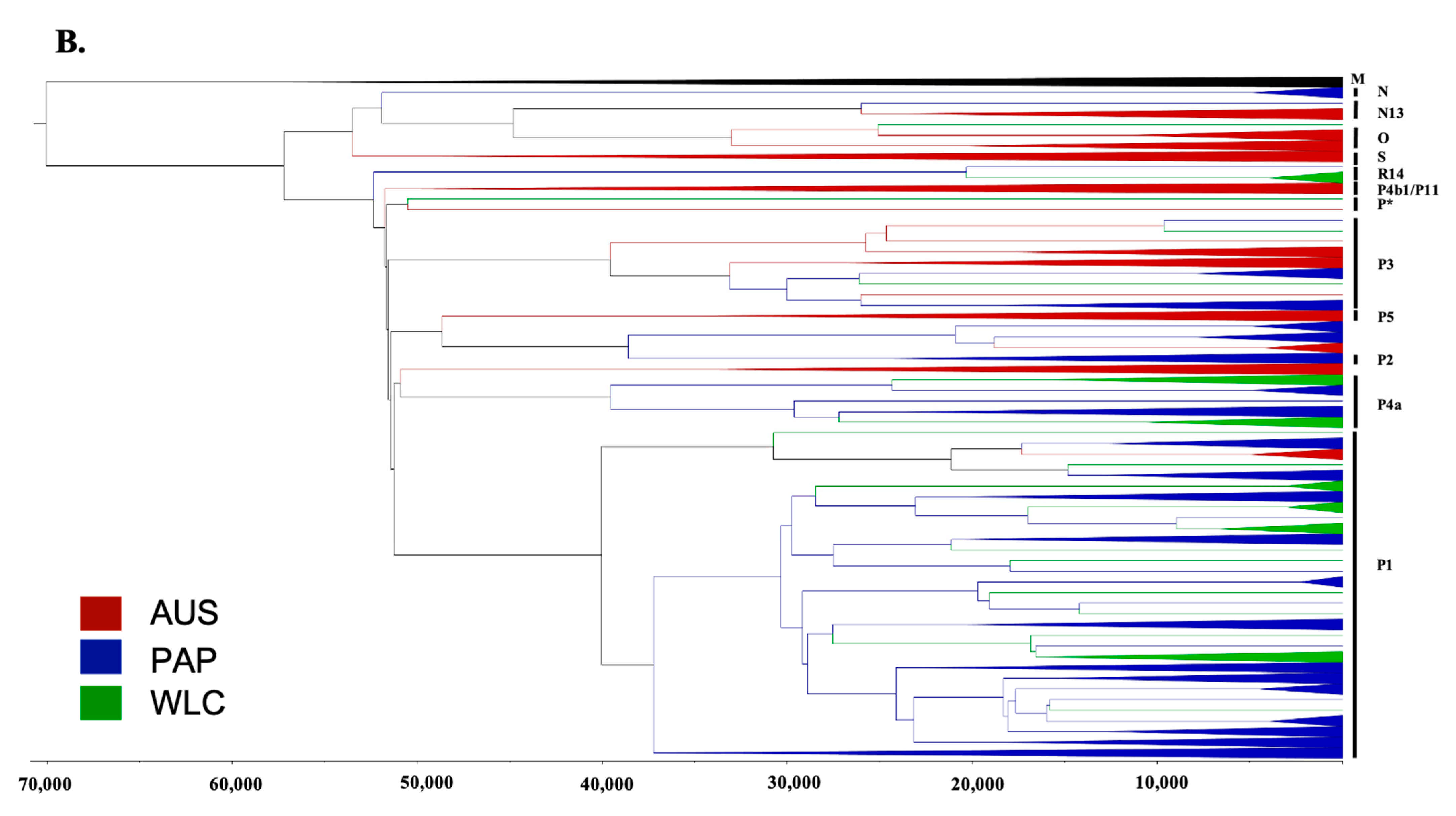
3.2. Phylogeographic Analyses
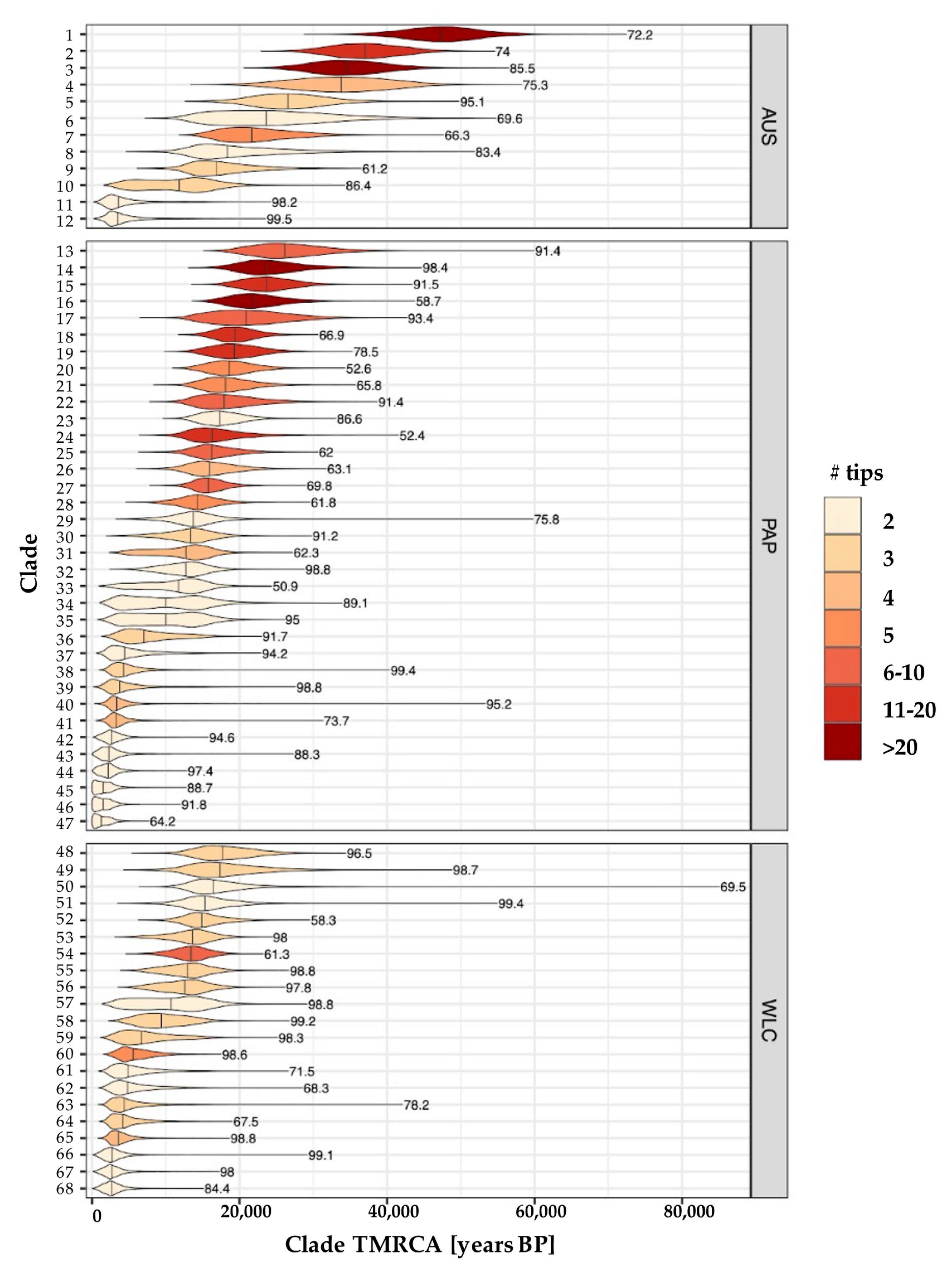
3.3. Estimating Rates and the Timing of Historical Movements
4. Discussion
4.1. Post-LGM Population Expansions and Movements
4.2. Redistribution of Papuan mtDNA Lineages Following Austronesian Contact
4.3. Comparison with Wallacean Archaeological and Linguistic Records
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergström, A.; Stringer, C.; Hajdinjak, M.; Scerri, E.M.L.; Skoglund, P. Origins of modern human ancestry. Nature 2021, 590, 229–237. [Google Scholar] [CrossRef]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into Human Genetic Variation and Population History from 929 Diverse Genomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 Genomes from 142 Diverse Populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef]
- Pedro, N.; Brucato, N.; Fernandes, V.; André, M.; Saag, L.; Pomat, W.; Besse, C.; Boland, A.; Deleuze, J.-F.; Clarkson, C.; et al. Papuan Mitochondrial Genomes and the Settlement of Sahul. J. Hum. Genet. 2020, 65, 875–887. [Google Scholar] [CrossRef]
- Tobler, R.; Rohrlach, A.; Soubrier, J.; Bover, P.; Llamas, B.; Tuke, J.; Bean, N.; Abdullah-Highfold, A.; Agius, S.; O’Donoghue, A.; et al. Aboriginal Mitogenomes Reveal 50,000 Years of Regionalism in Australia. Nature 2017, 544, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Malaspinas, A.-S.; Westaway, M.C.; Muller, C.; Sousa, V.C.; Lao, O.; Alves, I.; Bergström, A.; Athanasiadis, G.; Cheng, J.Y.; Crawford, J.E.; et al. A Genomic History of Aboriginal Australia. Nature 2016, 538, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Bergström, A.; Oppenheimer, S.J.; Mentzer, A.J.; Auckland, K.; Robson, K.; Attenborough, R.; Alpers, M.P.; Koki, G.; Pomat, W.; Siba, P.; et al. A Neolithic Expansion, but Strong Genetic Structure, in the Independent History of New Guinea. Science 2017, 357, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; O’Connell, J. Both Half right:Updating the Evidence for Dating First Human Arrivals in Sahul. Aust. Archaeol. 2014, 79, 86–108. [Google Scholar] [CrossRef]
- O’Connell, J.F.; Allen, J.; Williams, M.A.J.; Williams, A.N.; Turney, C.S.M.; Spooner, N.A.; Kamminga, J.; Brown, G.; Cooper, A. When Did Homo Sapiens First Reach Southeast Asia and Sahul? Proc. Natl. Acad. Sci. USA 2018, 115, 8482–8490. [Google Scholar] [CrossRef]
- Clarkson, C.; Jacobs, Z.; Marwick, B.; Fullagar, R.; Wallis, L.; Smith, M.; Roberts, R.G.; Hayes, E.; Lowe, K.; Carah, X.; et al. Human Occupation of Northern Australia by 65,000 Years Ago. Nature 2017, 547, 306–310. [Google Scholar] [CrossRef]
- Balme, J. Of Boats and String: The Maritime Colonisation of Australia. Quat. Int. 2013, 285, 68–75. [Google Scholar] [CrossRef]
- Brumm, A.; van den Bergh, G.D.; Storey, M.; Kurniawan, I.; Alloway, B.V.; Setiawan, R.; Setiyabudi, E.; Grün, R.; Moore, M.W.; Yurnaldi, D.; et al. Age and Context of the Oldest Known Hominin Fossils from Flores. Nature 2016, 534, 249–253. [Google Scholar] [CrossRef]
- Kealy, S.; Louys, J.; O’Connor, S. Reconstructing palaeogeography and inter-island visibility in the Wallacean archipelago during the likely period of Sahul colonization, 65-45 000 years ago: Reconstructing palaeogeography and intervisibility in Wallacea. Archaeol. Prospect. 2017, 24, 259–272. [Google Scholar] [CrossRef]
- Kealy, S.; Louys, J.; O’Connor, S. Least-cost pathway models indicate northern human dispersal from Sunda to Sahul. J. Hum. Evol. 2018, 125, 59–70. [Google Scholar] [CrossRef]
- Bird, M.I.; Beaman, R.J.; Condie, S.A.; Cooper, A.; Ulm, S.; Veth, P. Palaeogeography and voyage modeling indicates early human colonization of Australia was likely from Timor-Roti. Quat. Sci. Rev. 2018, 191, 431–439. [Google Scholar] [CrossRef]
- Norman, K.; Inglis, J.; Clarkson, C.; Faith, J.T.; Shulmeister, J.; Harris, D. An early colonisation pathway into northwest Australia 70–60,000 years ago. Quat. Sci. Rev. 2018, 180, 229–239. [Google Scholar] [CrossRef]
- Kealy, S.; Louys, J.; O’Connor, S. Islands under the Sea: A Review of Early Modern Human Dispersal Routes and Migration Hypotheses Through Wallacea. J. Isl. Coast. Archaeol. 2016, 11, 364–384. [Google Scholar] [CrossRef]
- Brumm, A.; Oktaviana, A.A.; Burhan, B.; Hakim, B.; Lebe, R.; Zhao, J.-X.; Sulistyarto, P.H.; Ririmasse, M.; Adhityatama, S.; Sumantri, I.; et al. Oldest Cave Art Found in Sulawesi. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Shipton, C.; O’Connor, S.; Jankowski, N.; O’Connor-Veth, J.; Maloney, T.; Kealy, S.; Boulanger, C. A New 44,000-Year Sequence from Asitau Kuru (Jerimalai), Timor-Leste, Indicates Long-Term Continuity in Human Behaviour. Archaeol. Anthropol. Sci. 2019, 11, 5717–5741. [Google Scholar] [CrossRef]
- Hawkins, S.; O’Connor, S.; Maloney, T.R.; Litster, M.; Kealy, S.; Fenner, J.N.; Aplin, K.; Boulanger, C.; Brockwell, S.; Willan, R.; et al. Oldest human occupation of Wallacea at Laili Cave, Timor-Leste, shows broad-spectrum foraging responses to late Pleistocene environments. Quat. Sci. Rev. 2017, 171, 58–72. [Google Scholar] [CrossRef]
- Sutikna, T.; Tocheri, M.W.; Faith, J.T.; Jatmiko; Due Awe, R.; Meijer, H.J.M.; Wahyu Saptomo, E.; Roberts, R.G. The Spatio-Temporal Distribution of Archaeological and Faunal Finds at Liang Bua (Flores, Indonesia) in Light of the Revised Chronology for Homo Floresiensis. J. Hum. Evol. 2018, 124, 52–74. [Google Scholar] [CrossRef]
- Friedlaender, J.S.; Friedlaender, F.R.; Hodgson, J.A.; Stoltz, M.; Koki, G.; Horvat, G.; Zhadanov, S.; Schurr, T.G.; Merriwether, D.A. Melanesian mtDNA Complexity. PLoS ONE 2007, 2, e248. [Google Scholar] [CrossRef]
- Hill, C.; Soares, P.; Mormina, M.; Macaulay, V.; Clarke, D.; Blumbach, P.B.; Vizuete-Forster, M.; Forster, P.; Bulbeck, D.; Oppenheimer, S.; et al. A Mitochondrial Stratigraphy for Island Southeast Asia. Am. J. Hum. Genet. 2007, 80, 29–43. [Google Scholar] [CrossRef]
- Tumonggor, M.K.; Karafet, T.M.; Hallmark, B.; Lansing, J.S.; Sudoyo, H.; Hammer, M.F.; Cox, M.P. The Indonesian Archipelago: An Ancient Genetic Highway Linking Asia and the Pacific. J. Hum. Genet. 2013, 58, 165–173. [Google Scholar] [CrossRef]
- HUGO Pan-Asian SNP Consortium; Abdulla, M.A.; Ahmed, I.; Assawamakin, A.; Bhak, J.; Brahmachari, S.K.; Calacal, G.C.; Chaurasia, A.; Chen, C.-H.; Chen, J.; et al. Mapping Human Genetic Diversity in Asia. Science 2009, 326, 1541–1545. [Google Scholar]
- Lipson, M.; Loh, P.-R.; Patterson, N.; Moorjani, P.; Ko, Y.-C.; Stoneking, M.; Berger, B.; Reich, D. Reconstructing Austronesian Population History in Island Southeast Asia. Nat. Commun. 2014, 5, 4689. [Google Scholar] [CrossRef]
- Hudjashov, G.; Karafet, T.M.; Lawson, D.J.; Downey, S.; Savina, O.; Sudoyo, H.; Lansing, J.S.; Hammer, M.F.; Cox, M.P. Complex Patterns of Admixture across the Indonesian Archipelago. Mol. Biol. Evol. 2017, 34, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. Available online: http://arxiv.org/abs/1303.3997 (accessed on 16 March 2013).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. Available online: http://arxiv.org/abs/1207.3907 (accessed on 17 July 2020).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstätter, A.; Forer, L.; Specht, G.; Bandelt, H.-J.; Kronenberg, F.; Salas, A.; Schönherr, S. HaploGrep 2: Mitochondrial Haplogroup Classification in the Era of High-Throughput Sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef]
- Van Oven, M. PhyloTree Build 17: Growing the human mitochondrial DNA tree. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e392–e394. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. Trimal: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Bouckaert, R.R.; Drummond, A.J. Bmodeltest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef]
- Heupink, T.H.; Subramanian, S.; Wright, J.L.; Endicott, P.; Westaway, M.C.; Huynen, L.; Parson, W.; Millar, C.D.; Willerslev, E.; Lambert, D.M. Ancient mtDNA Sequences from the First Australians Revisited. Proc. Natl. Acad. Sci. USA 2016, 113, 6892–6897. [Google Scholar] [CrossRef]
- Posth, C.; Renaud, G.; Mittnik, A.; Drucker, D.G.; Rougier, H.; Cupillard, C.; Valentin, F.; Thevenet, C.; Furtwängler, A.; Wißing, C.; et al. Pleistocene Mitochondrial Genomes Suggest a Single Major Dispersal of Non-Africans and a Late Glacial Population Turnover in Europe. Curr. Biol. 2016, 26, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Heled, J.; Bouckaert, R.R. Looking for Trees in the Forest: Summary Tree from Posterior Samples. BMC Evol. Biol. 2013, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Orme, D.; Freckleton, R.; Thomas, G.; Petzoldt, T.; Fritz, S.; Isaac, N.; Pearse, W. The Caper Package: Comparative Analysis of Phylogenetics and Evolution in R. R Package Version 2013, 5, 1–36. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 24 November 2019).
- Lewis, S.E.; Sloss, C.R.; Murray-Wallace, C.V.; Woodroffe, C.D.; Smithers, S.G. Post-glacial sea-level changes around the Australian margin: A review. Quat. Sci. Rev. 2013, 74, 115–138. [Google Scholar] [CrossRef]
- Pagel, M.; Meade, A. BayesTraits. 2007. Available online: http://www.evolutionrdgacuk/BayesTraitshtml (accessed on 24 November 2019).
- Xie, W.; Lewis, P.O.; Fan, Y.; Kuo, L.; Chen, M.-H. Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Syst. Biol. 2011, 60, 150–160. [Google Scholar] [CrossRef]
- Gilks, W.R.; Richardson, S.; Spiegelhalter, D.J. Introducing Markov chain Monte. Markov Chain Monte Carlo Pract. 1995, 1. [Google Scholar]
- Bollback, J.P. SIMMAP: Stochastic character mapping of discrete traits on phylogenies. BMC Bioinform. 2006, 7, 88. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Kong, Q.-P.; Sun, C.; Wang, H.-W.; Zhao, M.; Wang, W.-Z.; Zhong, L.; Hao, X.-D.; Pan, H.; Wang, S.-Y.; Cheng, Y.-T.; et al. Large-Scale mtDNA Screening Reveals a Surprising Matrilineal Complexity in East Asia and Its Implications to the Peopling of the Region. Mol. Biol. Evol. 2011, 28, 513–522. [Google Scholar] [CrossRef]
- Ricaut, F.-X.; Razafindrazaka, H.; Cox, M.P.; Dugoujon, J.-M.; Guitard, E.; Sambo, C.; Mormina, M.; Mirazon-Lahr, M.; Ludes, B.; Crubézy, E. A New Deep Branch of Eurasian mtDNA Macrohaplogroup M Reveals Additional Complexity Regarding the Settlement of Madagascar. BMC Genom. 2009, 10, 605. [Google Scholar] [CrossRef]
- Fregel, R.; Cabrera, V.; Larruga, J.M.; Abu-Amero, K.K.; González, A.M. Carriers of Mitochondrial DNA Macrohaplogroup N Lineages Reached Australia around 50,000 Years Ago following a Northern Asian Route. PLoS ONE 2015, 10, e0129839. [Google Scholar] [CrossRef]
- Clark, P.U.; Dyke, A.S.; Shakun, J.D.; Carlson, A.E.; Clark, J.; Wohlfarth, B.; Mitrovica, J.X.; Hostetler, S.W.; McCabe, A.M. The Last Glacial Maximum. Science 2009, 325, 710–714. [Google Scholar] [CrossRef]
- Diamond, J.; Bellwood, P. Farmers and their languages: The first expansions. Science 2003, 300, 597–603. [Google Scholar] [CrossRef]
- Denham, T.; Barton, H. A model of continuity from pre-existing foraging practices. Behav. Ecol. Transit. Agric. 2006, 1, 237. [Google Scholar]
- Reeves, J.M.; Barrows, T.T.; Cohen, T.J.; Kiem, A.S.; Bostock, H.C.; Fitzsimmons, K.E.; Jansen, J.D.; Kemp, J.; Krause, C.; Petherick, L.; et al. Climate Variability over the Last 35,000 Years Recorded in Marine and Terrestrial Archives in the Australian Region: An OZ-INTIMATE Compilation. Quat. Sci. Rev. 2013, 74, 21–34. [Google Scholar] [CrossRef]
- Wicaksono, S.A.; Russell, J.M.; Holbourn, A.; Kuhnt, W. Hydrological and vegetation shifts in the Wallacean region of central Indonesia since the Last Glacial Maximum. Quat. Sci. Rev. 2017, 157, 152–163. [Google Scholar] [CrossRef]
- Larena, M.; Sanchez-Quinto, F.; Sjödin, P.; McKenna, J.; Ebeo, C.; Reyes, R.; Casel, O.; Huang, J.-Y.; Hagada, K.P.; Guilay, D.; et al. Multiple migrations to the Philippines during the last 50,000 years. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Nagle, N.; van Oven, M.; Wilcox, S.; van Holst Pellekaan, S.; Tyler-Smith, C.; Xue, Y.; Ballantyne, K.N.; Wilcox, L.; Papac, L.; Cooke, K.; et al. Aboriginal Australian mitochondrial genome variation—An increased understanding of population antiquity and diversity. Sci. Rep. 2017, 7, 43041. [Google Scholar] [CrossRef]
- Gomes, S.M.; Bodner, M.; Souto, L.; Zimmermann, B.; Huber, G.; Strobl, C.; Röck, A.W.; Achilli, A.; Olivieri, A.; Torroni, A.; et al. Human Settlement History between Sunda and Sahul: A Focus on East Timor (Timor-Leste) and the Pleistocenic mtDNA Diversity. BMC Genom. 2015, 16, 70. [Google Scholar] [CrossRef]
- Stephen Lansing, J.; Cox, M.P.; de Vet, T.A.; Downey, S.S.; Hallmark, B.; Sudoyo, H. An ongoing Austronesian expansion in Island Southeast Asia. J. Anthropol. Archaeol. 2011, 30, 262–272. [Google Scholar] [CrossRef]
- Shipton, C.; O’Connor, S.; Kealy, S. The biogeographic threshold of Wallacea in human evolution. Quat. Int. 2021, 574, 1–12. [Google Scholar] [CrossRef]
- O’Connor, S.; Ono, R.; Clarkson, C. Pelagic fishing at 42,000 years before the present and the maritime skills of modern humans. Science 2011, 334, 1117–1121. [Google Scholar] [CrossRef]
- McClean, N. Conservation across the Cultural Divide: Comparative Perspectives on Local Communities and Protected Area Management in the Asia-Pacific Region; ANU College of Asia and the Pacific, Australian National University: Canberra, Australia, 2015. [Google Scholar]
- Reepmeyer, C.; O’Connor, S.; Mahirta; Kealy, S.; Maloney, T. Kisar, a small island participant in an extensive maritime obsidian network in the Wallacean Archipelago. Archaeol. Res. Asia 2019, 19, 100139. [Google Scholar] [CrossRef]
- Reepmeyer, C.; O’Connor, S.; Mahirta; Maloney, T.; Kealy, S. Late Pleistocene/early Holocene maritime interaction in Southeastern Indonesia—Timor Leste. J. Archaeol. Sci. 2016, 76, 21–30. [Google Scholar] [CrossRef]
- Kealy, S.; O’Connor, S.; Sari, D.M.; Shipton, C.; Langley, M.C.; Boulanger, C.; Kaharudin, H.A.F.; Patridina, E.P.B.G.G.; Algifary, M.A.; Irfan, A.; et al. Forty-thousand years of maritime subsistence near a changing shoreline on Alor Island (Indonesia). Quat. Sci. Rev. 2020, 249, 106599. [Google Scholar] [CrossRef]
- Bellwood, P. The Spice Islands in PrehistoryArchaeology in the Northern Moluccas, Indonesia; ANU Press: Canberra, Australia, 2019. [Google Scholar]
- Kaharudin, H.A.F.; Alifah, A.; Ramadhan, L.; Kealy, S. A review of archaeological dating efforts at cave and rockshelter sites in the Indonesian Archipelago. J. Indo Pac. Archaeol. 2020, 44, 80–112. [Google Scholar]
- Shipton, C.; O’Connor, S.; Kealy, S.; Mahirta; Syarqiyah, I.N.; Alamsyah, N.; Ririmasse, M. Early ground axe technology in Wallacea: The first excavations on Obi Island. PLoS ONE 2020, 15, e0236719. [Google Scholar] [CrossRef]
- Bellwood, P.; Irwin, G.; Tanudirjo, D. Lithic and other non-ceramic artefacts. In The Spice Islands in Prehistory Archaeology in the Northern Moluccas, Indonesia; Bellwood, P., Ed.; ANU Press: Canberra, Australia, 2019; pp. 107–120. [Google Scholar]
- Bellwood, P.; Irwin, G.; Tanudirjo, D.; Nitihaminoto, G.; Siswanto, J.; Bowdery, D. Investigations on Gebe Island. In The Spice Islands in Prehistory: Archaeology in the Northern Moluccas, Indonesia; Bellwood, P., Ed.; ANU Press: Canberra, Australia, 2019; pp. 15–44. [Google Scholar]
- Galipaud, J.-C.; Kinaston, R.; Halcrow, S.; Foster, A.; Harris, N.; Simanjuntak, T.; Javelle, J.; Buckley, H. The Pain Haka burial ground on Flores: Indonesian evidence for a shared Neolithic belief system in Southeast Asia. Antiquity 2016, 90, 1505–1521. [Google Scholar] [CrossRef]
- Szabó, K. Worked shell from the Northern Moluccas. In The Spice Islands in Prehistory: Archaeology in the Northern Moluccas, Indonesia; ANU Press: Canberra, Australia, 2019; pp. 121–134. [Google Scholar] [CrossRef]
- Smith, A.; Allen, J.; Hall, J.; McNiven, I. Pleistocene shell technologies: Evidence from Island Melanesia. Aust. Coast. Archaeol. 1999, 31, 291–297. [Google Scholar]
- Szabo, K.A. Shell Artefacts and Shell-Working within the Lapita Cultural Complex. 2010. Available online: http://ro.uow.edu.au/cgi/viewcontent.cgi?article=1533&context=scipapers (accessed on 17 July 2020).
- Valentin, F.; Spriggs, M.; Bedford, S.; Buckley, H. Vanuatu mortuary practices over three millennia: Lapita to the early European contact period. J. Pac. Archaeol. 2011, 2, 49–65. [Google Scholar]
- Ono, R.; Aziz, F.; Oktaviana, A.A.; Prastiningtyas, D.; Ririmasse, M.; Iriyanto, N.; Zesse, I.; Hisa, Y.; Yoneda, M. Development of Regional Maritime Networks during the Early Metal Age in Northern Maluku Islands: A View from Excavated Glass Ornaments and Pottery Variation. J. Island Coast. Archaeol. 2018, 13, 90–108. [Google Scholar] [CrossRef]
- Wisseman, J.C. Ian Glover. Archaeology in East Timor, 1966–1967. [Terra Australis 11.] xxii 241 pages, 102 illustrations, 132 tables. 1986. Canberra: Department of Prehistory, Research School of Pacific Studies, Australian National University; ISBN 0-86784-943-6 paperback Aus$18.50. Antiquity 1988, 399–400. [Google Scholar] [CrossRef]
- O’Connor, S. Rethinking the Neolithic in Island Southeast Asia, with Particular Reference to the Archaeology of Timor. Archipel 2015, 15–47. [Google Scholar] [CrossRef]
- Kealy, S.; Donnellan, S.C.; Mitchell, K.J.; Herrera, M.; Aplin, K.; O’Connor, S.; Louys, J. Phylogenetic Relationships of the Cuscuses (Diprotodontia: Phalangeridae) of Island Southeast Asia and Melanesia Based on the Mitochondrial ND2 Gene. Aust. Mammal. 2020, 42, 266. [Google Scholar] [CrossRef]
- Schapper, A. Finding Bunaq: The homeland and expansion of the Bunaq in central Timor. Life Land Timor Ethnogr. Pap. 2011, 163–186. [Google Scholar]
- McWilliam, A. Austronesians in Linguistic Disguise: Fataluku Cultural Fusion in East Timor. J. Southeast Asian Stud. 2007, 38, 355–375. [Google Scholar] [CrossRef]
- Hajek, J.; Himmelmann, N.; Bowden, J. Lóvaia: An East Timorese language on the verge of extinction. Int. J. Soc. Lang. 2003, 155–167. [Google Scholar] [CrossRef]
- O’Connor, S. Nine New Painted Rock Art Sites from East Timor in the Context of the Western Pacific Region. Asian Perspect. 2003, 42, 96–128. [Google Scholar] [CrossRef]
- Teixeira, J.C.; Cooper, A. Using hominin introgression to trace modern human dispersals. Proc. Natl. Acad. Sci. USA 2019, 116, 15327–15332. [Google Scholar] [CrossRef]
- Choin, J.; Mendoza-Revilla, J.; Arauna, L.R.; Cuadros-Espinoza, S.; Cassar, O.; Larena, M.; Ko, A.M.-S.; Harmant, C.; Laurent, R.; Verdu, P.; et al. Genomic Insights into Population History and Biological Adaptation in Oceania. Nature 2021. [Google Scholar] [CrossRef]
- Jacobs, G.S.; Hudjashov, G.; Saag, L.; Kusuma, P.; Darusallam, C.C.; Lawson, D.J.; Mondal, M.; Pagani, L.; Ricaut, F.-X.; Stoneking, M.; et al. Multiple Deeply Divergent Denisovan Ancestries in Papuans. Cell 2019, 177, 1010–1021.e32. [Google Scholar] [CrossRef]
- Teixeira, J.C.; Jacobs, G.S.; Stringer, C.; Tuke, J.; Hudjashov, G.; Purnomo, G.A.; Sudoyo, H.; Cox, M.P.; Tobler, R.; Turney, C.S.M.; et al. Widespread Denisovan ancestry in Island Southeast Asia but no evidence of substantial super-archaic hominin admixture. Nat. Ecol. Evol. 2021, 5, 616–624. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purnomo, G.A.; Mitchell, K.J.; O’Connor, S.; Kealy, S.; Taufik, L.; Schiller, S.; Rohrlach, A.; Cooper, A.; Llamas, B.; Sudoyo, H.; et al. Mitogenomes Reveal Two Major Influxes of Papuan Ancestry across Wallacea Following the Last Glacial Maximum and Austronesian Contact. Genes 2021, 12, 965. https://doi.org/10.3390/genes12070965
Purnomo GA, Mitchell KJ, O’Connor S, Kealy S, Taufik L, Schiller S, Rohrlach A, Cooper A, Llamas B, Sudoyo H, et al. Mitogenomes Reveal Two Major Influxes of Papuan Ancestry across Wallacea Following the Last Glacial Maximum and Austronesian Contact. Genes. 2021; 12(7):965. https://doi.org/10.3390/genes12070965
Chicago/Turabian StylePurnomo, Gludhug A., Kieren J. Mitchell, Sue O’Connor, Shimona Kealy, Leonard Taufik, Sophie Schiller, Adam Rohrlach, Alan Cooper, Bastien Llamas, Herawati Sudoyo, and et al. 2021. "Mitogenomes Reveal Two Major Influxes of Papuan Ancestry across Wallacea Following the Last Glacial Maximum and Austronesian Contact" Genes 12, no. 7: 965. https://doi.org/10.3390/genes12070965
APA StylePurnomo, G. A., Mitchell, K. J., O’Connor, S., Kealy, S., Taufik, L., Schiller, S., Rohrlach, A., Cooper, A., Llamas, B., Sudoyo, H., Teixeira, J. C., & Tobler, R. (2021). Mitogenomes Reveal Two Major Influxes of Papuan Ancestry across Wallacea Following the Last Glacial Maximum and Austronesian Contact. Genes, 12(7), 965. https://doi.org/10.3390/genes12070965