
Angiotensin-Converting Enzyme 2 (ACE2) as a Potential Diagnostic and Prognostic Biomarker for Chronic Inflammatory Lung Diseases



{kind=link}
{kind=link}
Abstract
:1. Introduction
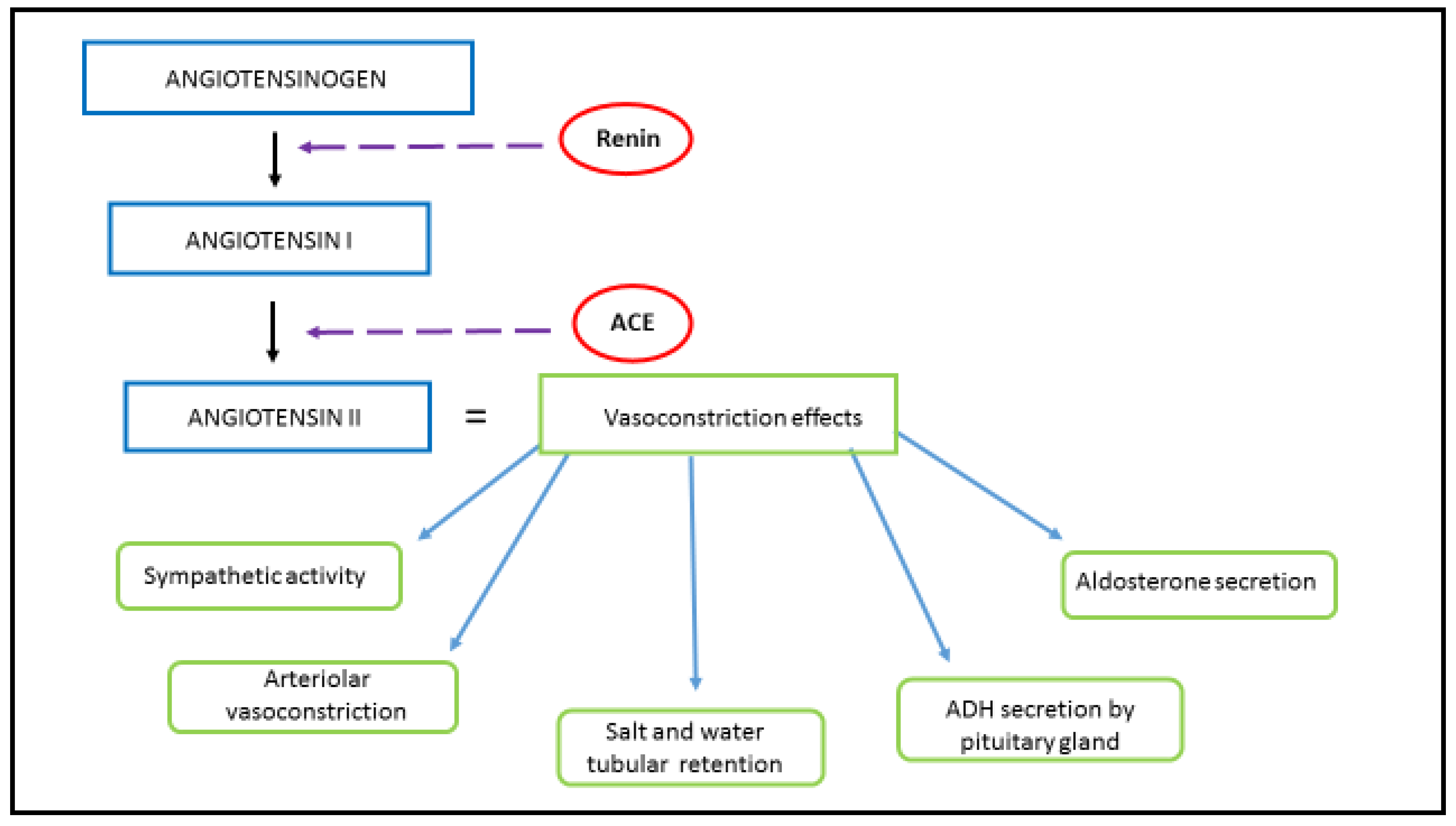
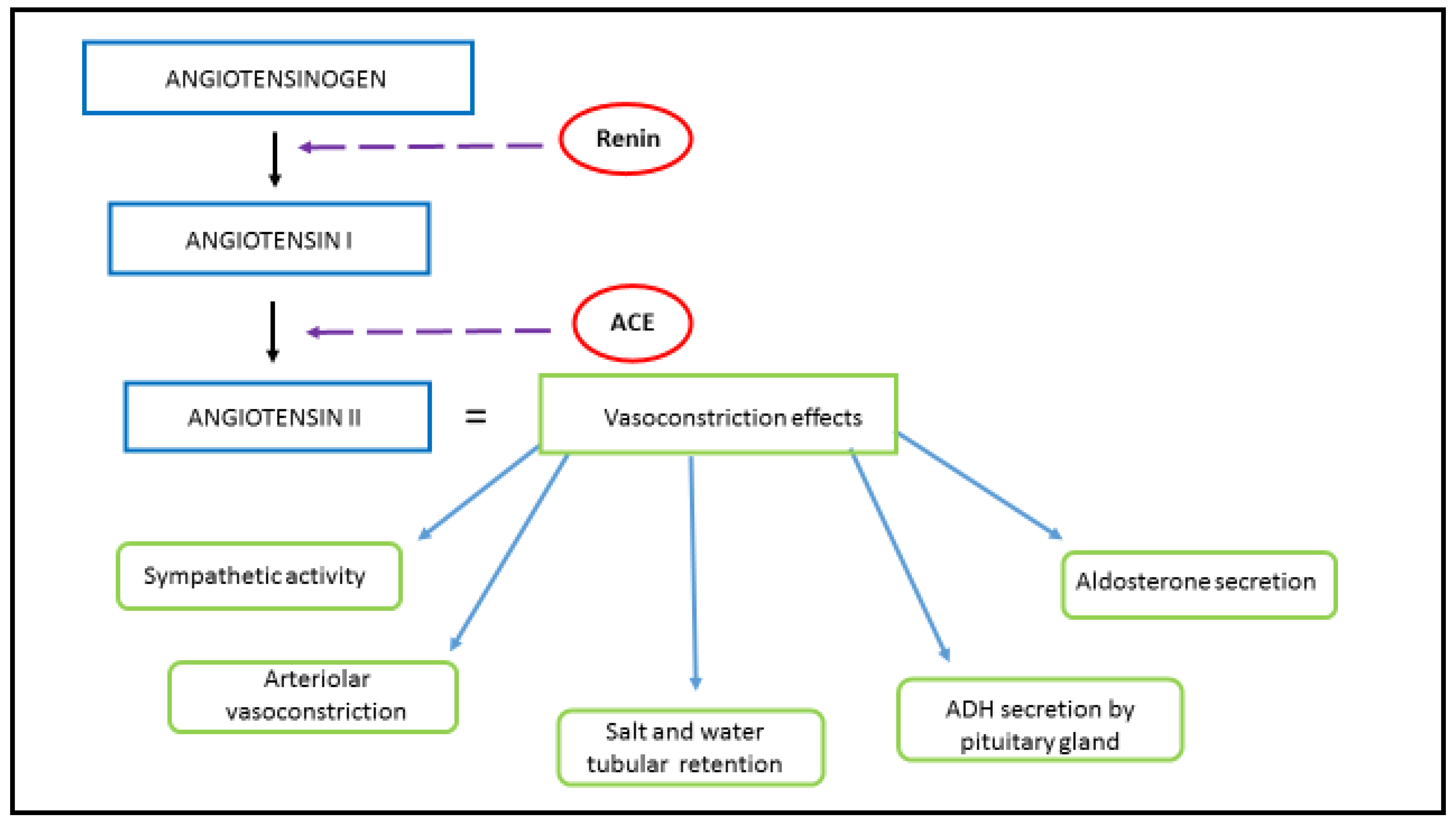
1.1. Physiological Function and Signaling Pathway of RAAS
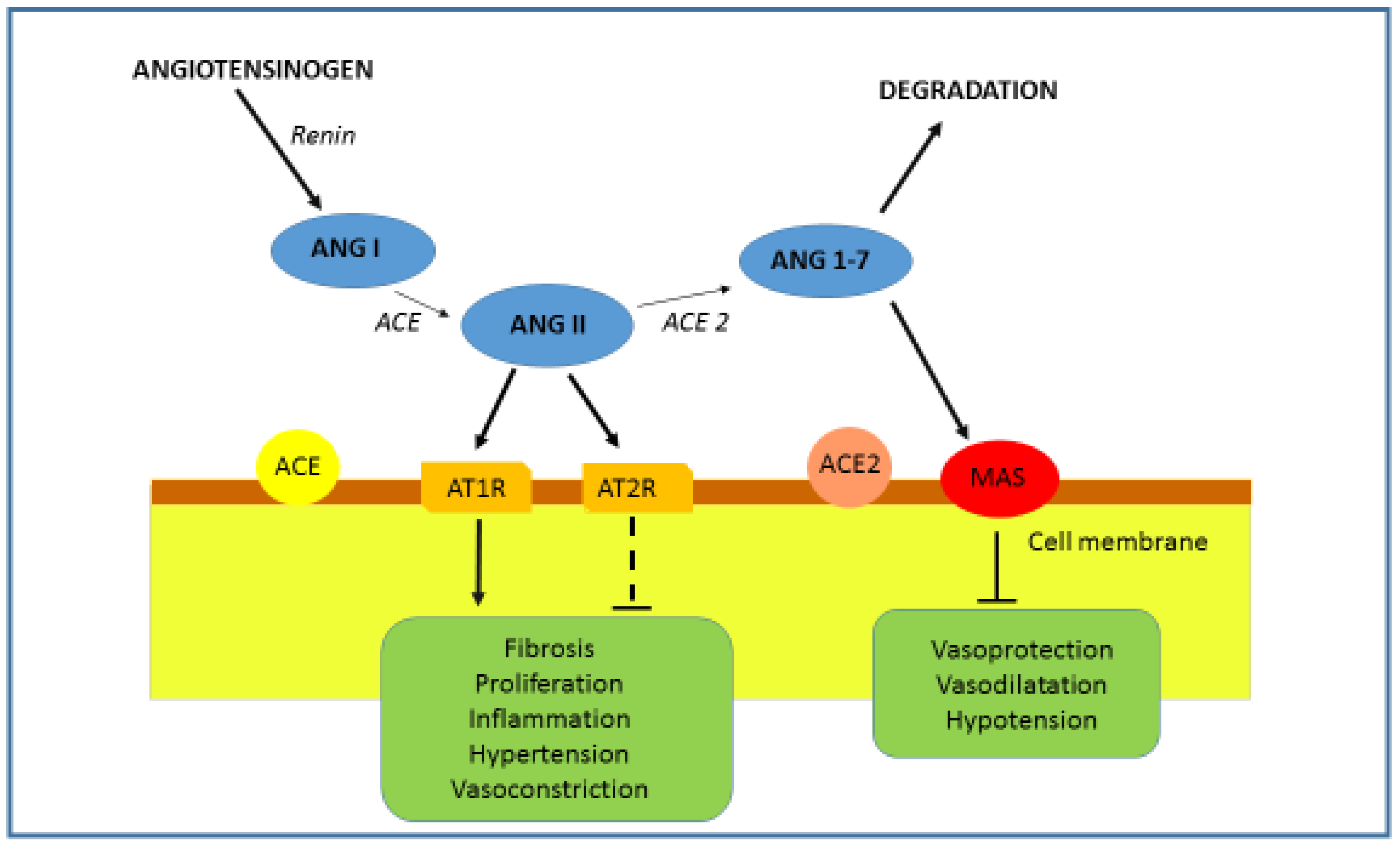
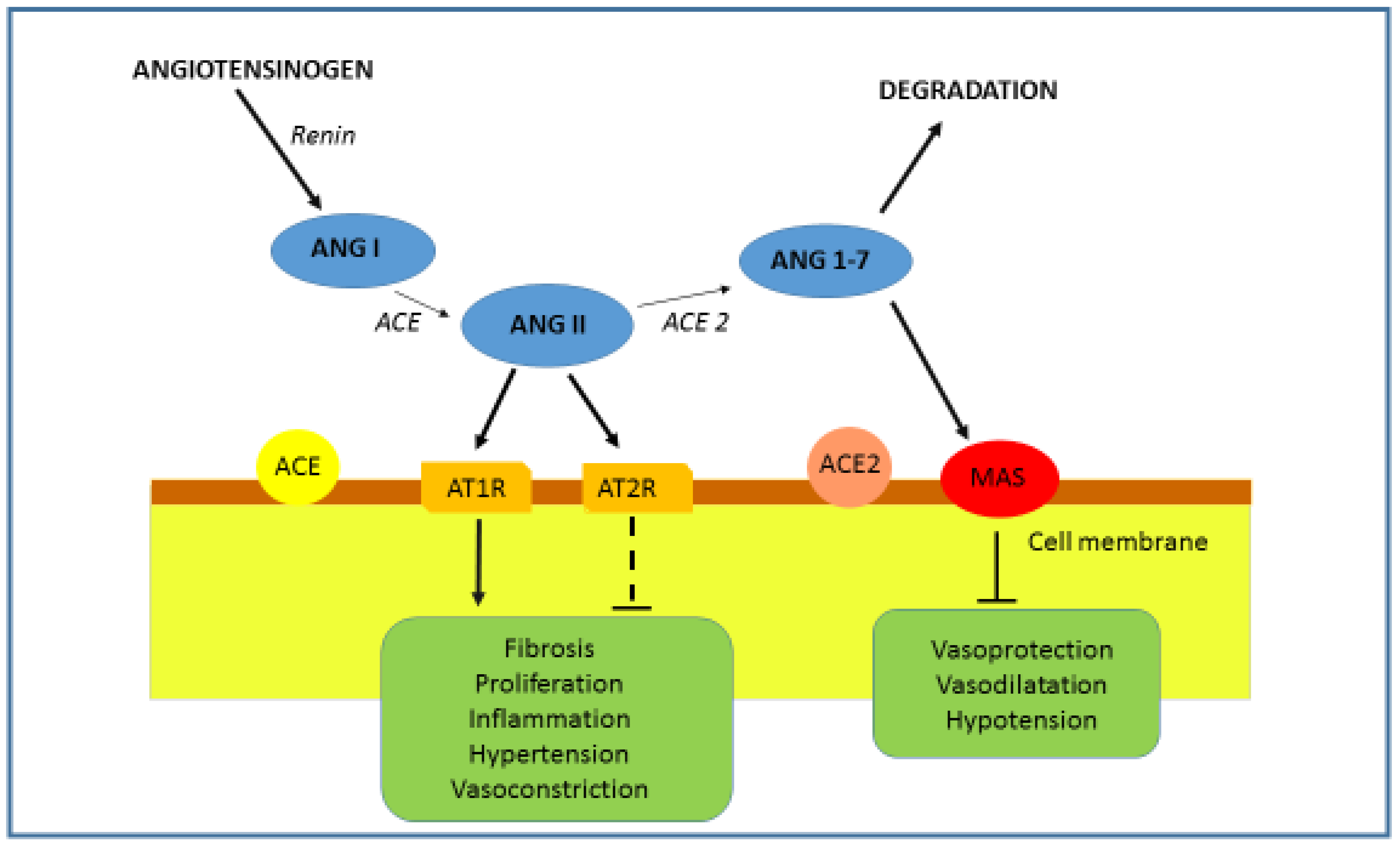
1.2. ACE2 and RAAS Regulation
1.3. ACE Inhibitors and ARBs
2. ACE2, Inflammation and Pulmonary Disease
3. COPD and ACE2
4. Asthma and ACE2
5. Pulmonary Hypertension and ACE2
6. COVID-19 and ACE2
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A Novel Angiotensin-Converting Enzyme—Related to Angiotensin 1-9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme: Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, M.E.; Thurman, A.; Pezzulo, A.A.; Leidinger, M.R.; Klesney-Tait, J.A.; Karp, P.H.; Tan, P.; Wohlford-Lenane, C.; McCray, P.B.; Meyerholz, D.K. Heterogeneous expression of the SARS-Coronavirus-2 receptor ACE2 in the human respiratory tract. EBioMedicine 2020, 60, 102976. [Google Scholar] [CrossRef] [PubMed]
- Harmer, D.; Gilbert, M.; Borman, R.; Clark, K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002, 532, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef]
- Fleming, I.; Kohlstedt, K.; Busse, R. The tissue renin-angiotensin system and intracellular signalling. Curr. Opin. Nephrol. Hypertens. 2006, 15, 8–13. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Lu, X.; Danser, A.H.J. Revisiting the Brain Renin-Angiotensin System—Focus on Novel Therapies. Curr. Hypertens. Rep. 2019, 21, 28. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santo, M.J. The ACE2/Angiotensin-(1-7)/Mas axis of the renin-angiotensin system: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikellis, C.; Thomas, M.C. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.M.E.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Crowley, S.D.; Gurley, S.B.; Herrera, M.J.; Ruiz, P.; Griffiths, R.; Kumar, A.P.; Kim, H.S.; Smithies, O.; Le, T.H.; Coffman, T.M. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc. Natl. Acad. Sci. USA 2006, 103, 17985–17990. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Bo, L.; Li, P.; Lu, X.; Li, W.; Pan, L.; Sun, Y.; Mu, D.; Liu, W.; Jin, F. Losartan, a selective antagonist of AT1 receptor, attenuates seawater inhalation induced lung injury via modulating JAK2/STATs and apoptosis in rat. Pulm. Pharmacol. Ther. 2017, 45, 69–79. [Google Scholar] [CrossRef]
- Wang, R.; Zagariya, A.; Ibarra-sunga, O.; Gidea, C.; Ang, E.; Deshmukh, S.; Chaudhary, G.; Baraboutis, J.; Filippatos, G.; Uhal, B.D. Angiotensin II induces apoptosis in human and rat alveolar epithelial cells. Am. J. Physiol. 1999, 276, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gao, Y.; Zhao, W.; Yu, G.; Jin, F. ACE-2/ANG1-7 ameliorates ER stress-induced apoptosis in seawater aspiration-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L1015–L1027. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.O.; Zanoni, F.L.; Martins, D.O.; Coimbra, R.; Krieger, J.E.; Jancar, S.; Sannomiya, P. Insulin regulates cytokines and intercellular adhesion molecule-1 gene expression through nuclear factor-κB activation in LPS-induced acute lung injury in rats. Shock 2009, 31, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.L.; Du, Y.; Zhang, C.; Cheng, C.; Yang, H.Y.; Jin, Y.F.; Duan, G.C.; Chen, S.Y. Role of renin-angiotensin system in acute lung injury caused by viral infection. Infect. Drug Resist. 2020, 13, 3715–3725. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef] [PubMed]
- Sarzani, R.; Giulietti, F.; Di Pentima, C.; Filipponi, A.; Spannella, F. Antagonizing the renin–angiotensin–aldosterone system in the era of COVID-19. Intern. Emerg. Med. 2020, 15, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Mo, H.; Cai, L.; Kong, T.; Zheng, W.; Ye, J.; Qi, J.; Xiao, Z. Losartan prevents sepsis-induced acute lung injury and decreases activation of nuclear factorκB and mitogen-activated protein kinases. Shock 2009, 31, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qiu, H.B.; Yang, Y.; Wang, L.; Ding, H.M.; Li, H.P. Losartan, an antagonist of AT1 receptor for angiotensin II, attenuates lipopolysaccharide-induced acute lung injury in rat. Arch. Biochem. Biophys. 2009, 481, 131–136. [Google Scholar] [CrossRef]
- Jenkins, T.A.; Chai, S.Y. Effect of chronic angiotensin converting enzyme inhibition on spatial memory and anxiety-like behaviours in rats. Neurobiol. Learn. Mem. 2007, 87, 218–224. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Brewster, U.C.; Perazella, M.A. The renin-angiotensin-aldosterone system and the kidney: Effects on kidney disease. Am. J. Med. 2004, 116, 263–272. [Google Scholar] [CrossRef]
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 23, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandivier, R.W.; Henson, P.M.; Douglas, I.S. Burying the dead: The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 2006, 129, 1673–1682. [Google Scholar] [CrossRef]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef]
- Ferreira, A.J.; Shenoy, V.; Qi, Y.; Fraga-Silva, R.A.; Santos, R.A.S.; Katovich, M.J.; Raizada, M.K. Angiotensin-converting enzyme 2 activation protects against hypertension-induced cardiac fibrosis involving extracellular signal-regulated kinases. Exp. Physiol. 2011, 96, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Yamazato, Y.; Ferreira, A.J.; Hong, K.H.; Sriramula, S.; Francis, J.; Yamazato, M.; Yuan, L.; Bradford, C.N.; Shenoy, V.; Oh, S.P.; et al. Prevention of pulmonary hypertension by angiotensin-converting enzyme 2 gene transfer. Hypertension 2009, 54, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Kuba, K.; Ohto-Nakanishi, T.; Penninger, J.M. Angiotensin-converting enzyme 2 (ACE2) in disease pathogenesis. Circ. J. 2010, 74, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suski, M.; Olszanecki, R.; Stachowicz, A.; Madej, J.; Bujak-Gizycka, B.; Okoń, K.; Korbut, R. The influence of angiotensin-(1-7) Mas receptor agonist (AVE 0991) on mitochondrial proteome in kidneys of apoE knockout mice. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 2463–2469. [Google Scholar] [CrossRef]
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.H.; Penninger, J.M.; Kassiri, Z.; et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122, 717–728. [Google Scholar] [CrossRef]
- Dorsainval, W. ACE2/Ang1-7 MAS AXIS: The counter-regulator of the classical renin angiotensin system. Mako NSU Undergrad. Stud. J. 2020, 2020, 2. [Google Scholar]
- Singh, D.; Agusti, A.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Criner, G.J.; Frith, P.; Halpin, D.M.G.; Han, M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: The GOLD science committee report 2019. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Soriano, J.B.; Abajobir, A.A.; Abate, K.H.; Abera, S.F.; Agrawal, A.; Ahmed, M.B.; Aichour, A.N.; Aichour, I.; Aichour, M.T.E.; Alam, K.; et al. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: A systematic analysis for the Global Burden of Disease Study. Lancet Respir. Med. 2017, 5, 691–706. [Google Scholar] [CrossRef] [Green Version]
- Bando, M.; Miyazawa, T.; Shinohara, H.; Owada, T.; Terakado, M.; Sugiyama, Y. An epidemiological study of the effects of statin use on airflow limitation in patients with chronic obstructive pulmonary disease. Respirology 2012, 17, 493–498. [Google Scholar] [CrossRef]
- Decramer, M.; Janssens, W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir. Med. 2013, 1, 73–83. [Google Scholar] [CrossRef]
- Scandiuzzi, L.; Beghdadi, W.; Daugas, E.; Åbrink, M.; Tiwari, N.; Brochetta, C.; Claver, J.; Arouche, N.; Zang, X.; Pretolani, M.; et al. Mouse Mast Cell Protease-4 Deteriorates Renal Function by Contributing to Inflammation and Fibrosis in Immune Complex-Mediated Glomerulonephritis. J. Immunol. 2010, 185, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Chao, J.; Wood, J.G.; Blanco, V.G.; Gonzalez, N.C. The systemic inflammation of alveolar hypoxia is initiated by alveolar macrophage-borne mediator(s). Am. J. Respir. Cell Mol. Biol. 2009, 41, 573–582. [Google Scholar] [CrossRef]
- Teramoto, S.; Suzuki, M.; Matsuse, T.; Ishii, T.; Fukuchi, Y.; Ouchi, Y. Effects of Angiotensin-Converting Enzyme Inhibitors on Spontaneous or Stimulated Generation of Reactive Oxygen Species by Bronchoalveolar Lavage Cells Harvested from Patients with or without Chronic Obstructive Pulmonary Disease. JPN J. Pharmacol. 2000, 83, 56–62. [Google Scholar] [CrossRef]
- Xue, T.; Wei, N.; Xin, Z.; Qingyu, X. Angiotensin-converting enzyme-2 overexpression attenuates inflammation in rat model of chronic obstructive pulmonary disease. Inhal. Toxicol. 2014, 26, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Finney, L.J.; Glanville, N.; Farne, H.; Aniscenko, J.; Fenwick, P.; Kemp, S.V.; Trujillo-Torralbo, M.B.; Loo, S.L.; Calderazzo, M.A.; Wedzicha, J.A.; et al. Inhaled corticosteroids downregulate the SARS-CoV-2 receptor ACE2 in COPD through suppression of type I interferon. J. Allergy Clin. Immunol. 2021, 147, 510–519.e5. [Google Scholar] [CrossRef]
- Oakes, J.M.; Fuchs, R.M.; Gardner, J.D.; Lazartigues, E.; Yue, X. Nicotine and the renin-angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R895–R906. [Google Scholar] [CrossRef] [Green Version]
- Vos, T.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; Aboyans, V.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Gelb, A.F.; Zamel, N.; Krishnan, A. Physiologic similarities and differences between asthma and chronic obstructive pulmonary disease. Curr. Opin. Pulm. Med. 2008, 14, 24–30. [Google Scholar] [CrossRef]
- Ravensberg, A.J.; Slats, A.M.; Van Wetering, S.; Janssen, K.; Van Wijngaarden, S.; De Jeu, R.; Rabe, K.F.; Sterk, P.J.; Hiemstra, P.S. CD8+ T cells characterize early smoking-related airway pathology in patients with asthma. Respir. Med. 2013, 107, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Fahy, J.V. Type 2 inflammation in asthma-present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Francisco, D.; Conway, M.; Martinez, F.D.; Vercelli, D.; Polverino, F.; Billheimer, D.; Kraft, M. Type 2 inflammation modulates ACE2 and TMPRSS2 in airway epithelial cells. J. Allergy Clin. Immunol. 2020, 146, 80–88.e8. [Google Scholar] [CrossRef]
- Song, J.; Zeng, M.; Wang, H.; Qin, C.; Hou, H.Y.; Sun, Z.Y.; Xu, S.P.; Wang, G.P.; Guo, C.L.; Deng, Y.K.; et al. Distinct effects of asthma and COPD comorbidity on disease expression and outcome in patients with COVID-19. Allergy Eur. J. Allergy Clin. Immunol. 2020, 76, 483–496. [Google Scholar] [CrossRef]
- Liu, S.; Zhi, Y.; Ying, S. COVID-19 and Asthma: Reflection During the Pandemic. Clin. Rev. Allergy Immunol. 2020, 59, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Sajuthi, S.P.; DeFord, P.; Li, Y.; Jackson, N.D.; Montgomery, M.T.; Everman, J.L.; Rios, C.L.; Pruesse, E.; Nolin, J.D.; Plender, E.G.; et al. Type 2 and interferon inflammation regulate SARS-CoV-2 entry factor expression in the airway epithelium. Nat. Commun. 2020, 11, 5139. [Google Scholar] [CrossRef] [PubMed]
- Brough, H.A.; Kalayci, O.; Sediva, A.; Untersmayr, E.; Munblit, D.; Rodriguez del Rio, P.; Vazquez-Ortiz, M.; Arasi, S.; Alvaro-Lozano, M.; Tsabouri, S.; et al. Managing childhood allergies and immunodeficiencies during respiratory virus epidemics—The 2020 COVID-19 pandemic: A statement from the EAACI-section on pediatrics. Pediatr. Allergy Immunol. 2020, 31, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.L. Asthma and COVID-19: Is asthma a risk factor for severe outcomes? Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 1543–1545. [Google Scholar] [CrossRef]
- Myou, S.; Fujimura, M.; Kurashima, K.; Tachibana, H.; Watanabe, K.; Hirose, T. Type 1 angiotensin II receptor antagonism reduces antigen-induced airway reactions. Am. J. Respir. Crit. Care Med. 2000, 162, 45–49. [Google Scholar] [CrossRef]
- Magalhães, G.S.; Rodrigues-Machado, M.G.; Motta-Santos, D.; Silva, A.R.; Caliari, M.V.; Prata, L.O.; Abreu, S.C.; Rocco, P.R.M.; Barcelos, L.S.; Santos, R.A.S.; et al. Angiotensin-(1-7) attenuates airway remodelling and hyperresponsiveness in a model of chronic allergic lung inflammation. Br. J. Pharmacol. 2015, 172, 2330–2342. [Google Scholar] [CrossRef] [Green Version]
- El-Hashim, A.Z.; Renno, W.M.; Raghupathy, R.; Abduo, H.T.; Akhtar, S.; Benter, I.F. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-kB-dependent pathways. Br. J. Pharmacol. 2012, 166, 1964–1976. [Google Scholar] [CrossRef] [Green Version]
- Bai, T.R.; Vonk, J.M.; Postma, D.S.; Boezen, H.M. Severe exacerbations predict excess lung function decline in asthma. Eur. Respir. J. 2007, 30, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Lemanske, R.F.; Busse, W.W. Asthma: Clinical expression and molecular mechanisms. J. Allergy Clin. Immunol. 2010, 125, S95–S102. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.C.; Sajuthi, S.; Deford, P.; Christenson, S.; Rios, C.L.; Montgomery, M.T.; Woodruff, P.G.; Mauger, D.T.; Erzurum, S.C.; Johansson, M.W.; et al. COVID-19-related genes in sputum cells in asthma: Relationship to demographic features and corticosteroids. Am. J. Respir. Crit. Care Med. 2020, 202, 83–90. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hautefort, A.; Girerd, B.; Montani, D.; Cohen-Kaminsky, S.; Price, L.; Lambrecht, B.N.; Humbert, M.; Perros, F. T-helper 17 cell polarization in pulmonary arterial hypertension. Chest 2015, 147, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Leopold, J.A. The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series). Pulm. Circ. 2014, 4, 200–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, V.; Kwon, K.C.; Rathinasabapathy, A.; Lin, S.; Jin, G.; Song, C.; Shil, P.; Nair, A.; Qi, Y.; Li, Q.; et al. Oral delivery of angiotensin-converting enzyme 2 and angiotensin-(1-7) bioencapsulated in plant cells attenuates pulmonary hypertension. Hypertension 2014, 64, 1248–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampl, V.; Herget, J.; Bíbová, J.; Banasová, A.; Husková, Z.; Vańourková, Z.; Jíchová, S.; Kujal, P.; Vernerová, Z.; Sadowski, J.; et al. Intrapulmonary activation of the angiotensin-converting enzyme type 2/angiotensin 1-7/g-protein-coupled mas receptor axis attenuates pulmonary hypertension in ren-2 transgenic rats exposed to chronic hypoxia. Physiol. Res. 2015, 64, 25–38. [Google Scholar] [CrossRef]
- Yuan, Y.M.; Luo, L.; Guo, Z.; Yang, M.; Ye, R.S.; Luo, C. Activation of renin-angiotensin-aldosterone system (RAAS) in the lung of smoking-induced pulmonary arterial hypertension (PAH) rats. JRAAS-J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 249–253. [Google Scholar] [CrossRef]
- Guignabert, C.; De Man, F.; Lombès, M. ACE2 as therapy for pulmonary arterial hypertension: The good outweighs the bad. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, J.; Martin, M.; He, M.; Gongol, B.; Marin, T.L.; Chen, L.; Shi, X.; Yin, Y.; Shang, F.; et al. AMP-activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 509–520. [Google Scholar] [CrossRef]
- Xiantian, X.; Ping, C.; Jingfang, W.; Jiannan, F.; Hui, Z.; Xuan, L.; Wu, Z.; Pei, H. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases from the Chinese Center for Disease Control and Prevention. JAMA J. Am. Med. Assoc. 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Liang, W.; Zhao, Y.; Liang, H.; Chen, Z.; Li, Y.; Liu, X.; Chen, R.; Tang, C.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: A nationwide analysis. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, R.; Liu, Z. ACE2 exhibits protective effects against LPS-induced acute lung injury in mice by inhibiting the LPS-TLR4 pathway. Exp. Mol. Pathol. 2020, 113, 104350. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, G.; Knox, K.; Roy, A. Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID-19. The COVID-19 resource centre is hosted on Elsevier Connect, the company’s public news and information. Gene Rep. 2020, 23, 101077. [Google Scholar] [CrossRef]
- Feng, Y.; Wan, H.; Liu, J.; Zhang, R.; Ma, Q.; Han, B.; Xiang, Y.; Che, J.; Cao, H.; Fei, X.; et al. The angiotensin-converting enzyme 2 in tumor growth and tumor-associated angiogenesis in non-small cell lung cancer. Oncol. Rep. 2010, 23, 941–948. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marčetić, D.; Samaržija, M.; Vukić Dugac, A.; Knežević, J. Angiotensin-Converting Enzyme 2 (ACE2) as a Potential Diagnostic and Prognostic Biomarker for Chronic Inflammatory Lung Diseases. Genes 2021, 12, 1054. https://doi.org/10.3390/genes12071054
Marčetić D, Samaržija M, Vukić Dugac A, Knežević J. Angiotensin-Converting Enzyme 2 (ACE2) as a Potential Diagnostic and Prognostic Biomarker for Chronic Inflammatory Lung Diseases. Genes. 2021; 12(7):1054. https://doi.org/10.3390/genes12071054
Chicago/Turabian StyleMarčetić, Dejan, Miroslav Samaržija, Andrea Vukić Dugac, and Jelena Knežević. 2021. "Angiotensin-Converting Enzyme 2 (ACE2) as a Potential Diagnostic and Prognostic Biomarker for Chronic Inflammatory Lung Diseases" Genes 12, no. 7: 1054. https://doi.org/10.3390/genes12071054
APA StyleMarčetić, D., Samaržija, M., Vukić Dugac, A., & Knežević, J. (2021). Angiotensin-Converting Enzyme 2 (ACE2) as a Potential Diagnostic and Prognostic Biomarker for Chronic Inflammatory Lung Diseases. Genes, 12(7), 1054. https://doi.org/10.3390/genes12071054