
ZEB2, the Mowat-Wilson Syndrome Transcription Factor: Confirmations, Novel Functions, and Continuing Surprises



Abstract
1. Discovery of ZEB2
2. ZEB2 and Mowat-Wilson Syndrome, and beyond
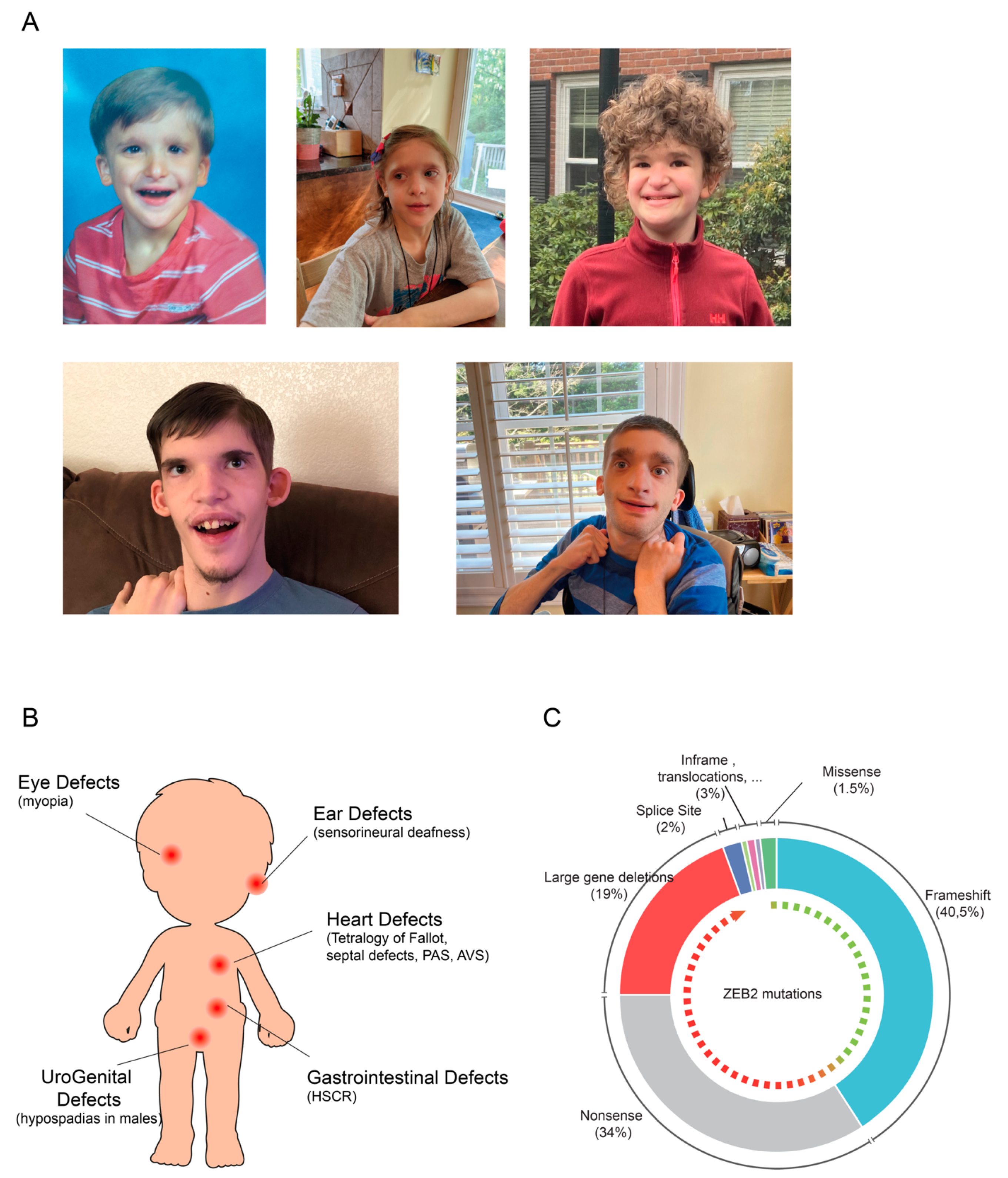
2.1. Mowat-Wilson Syndrome
2.2. ZEB2 Actions Studied Where Present
2.3. ZEB2 Levels Are Important, Too
3. ZEB2 Gene and Protein Organization
4. ZEB2 Regulation
5. ZEB2 in the Development of Nervous Systems in Vertebrates
5.1. Initial Studies in Xenopus Embryos
5.2. Modeling Neurodevelopmental Aspects of MOWS in Mice
5.2.1. General Zeb2-KO
5.2.2. MOWS and Neurodevelopmental Relevant Zeb2 Expression Domains in Mouse Embryos
5.2.3. Zeb2-cKO in the Neural Crest Cell Lineage
5.2.4. Neurocristopathies Reminiscent of MOWS
5.2.5. Dorsal Root Ganglia and Pain Sensing
5.2.6. The ENS and HSCR
5.2.7. Zeb2-cKO in the Developing Forebrain
5.2.8. ZEB2 in the Formation and Output of the Adult Neurogenic V-SVZ Compartment
5.2.9. ZEB2 in Midbrain, Hindbrain, and Spinal Cord
Midbrain
Hindbrain
Spinal Cord
5.2.10. Zeb2-cKO in Early and Late Retinogenesis, and Lens Formation
5.2.11. Zeb2-cKO in Embryonic Myelinogenesis and Postnatal (re)Myelination
6. Emerging Cellular Models for Studying ZEB2
7. General Conclusion and Future Perspectives
Author Contributions
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verschueren, K.; Remacle, J.E.; Collart, C.; Kraft, H.; Baker, B.S.; Tylzanowski, P.; Nelles, L.; Wuytens, G.; Su, M.T.; Bodmer, R.; et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5′-CACCT sequences in candidate target genes. J. Biol. Chem. 1999, 274, 20489–20498. [Google Scholar] [CrossRef]
- Funahashi, J.; Kamachi, Y.; Goto, K.; Kondoh, H. Identification of nuclear factor delta EF1 and its binding site essential for lens-specific activity of the delta 1-crystallin enhancer. Nucleic Acids Res. 1991, 19, 3543–3547. [Google Scholar] [CrossRef]
- Clark, S.G.; Chiu, C.C. elegans ZAG-1, a Zn-finger-homeodomain protein, regulates axonal development and neuronal differentiation. Development 2003, 130, 3781–3794. [Google Scholar] [CrossRef]
- Fortini, M.E.; Lai, Z.C.; Rubin, G.M. The Drosophila zfh-1 and zfh-2 genes encode novel proteins containing both zinc-finger and homeodomain motifs. Mech. Dev. 1991, 34, 113–122. [Google Scholar] [CrossRef]
- Liu, M.; Su, M.; Lyons, G.E.; Bodmer, R. Functional conservation of zinc-finger homeodomain gene zfh1/SIP1 in Drosophila heart development. Dev. Genes Evol. 2006, 216, 683–693. [Google Scholar] [CrossRef]
- Su, M.T.; Fujioka, M.; Goto, T.; Bodmer, R. The Drosophila homeobox genes zfh-1 and even-skipped are required for cardiac-specific differentiation of a numb-dependent lineage decision. Development 1999, 126, 3241–3251. [Google Scholar] [CrossRef]
- Wacker, I.; Schwarz, V.; Hedgecock, E.M.; Hutter, H. zag-1, a Zn-finger homeodomain transcription factor controlling neuronal differentiation and axon outgrowth in C. elegans. Development 2003, 130, 3795–3805. [Google Scholar] [CrossRef]
- Van de Putte, T.; Francis, A.; Nelles, L.; van Grunsven, L.A.; Huylebroeck, D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum. Mol. Genet. 2007, 16, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Lurie, I.W.; Supovitz, K.R.; Rosenblum-Vos, L.S.; Wulfsberg, E.A. Phenotypic variability of del(2) (q22–q23): Report of a case with a review of the literature. Genet. Couns. 1994, 5, 11–14. [Google Scholar] [PubMed]
- Cacheux, V.; Dastot-Le Moal, F.; Kääriäinen, H.; Bondurand, N.; Rintala, R.; Boissier, B.; Wilson, M.; Mowat, D.; Goossens, M. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum. Mol. Genet. 2001, 10, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Mowat, D.R.; Croaker, G.D.; Cass, D.T.; Kerr, B.A.; Chaitow, J.; Adès, L.C.; Chia, N.L.; Wilson, M.J. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: Delineation of a new syndrome and identification of a locus at chromosome 2q22–q23. J. Med. Genet. 1998, 35, 617–623. [Google Scholar] [CrossRef]
- Wakamatsu, N.; Yamada, Y.; Yamada, K.; Ono, T.; Nomura, N.; Taniguchi, H.; Kitoh, H.; Mutoh, N.; Yamanaka, T.; Mushiake, K.; et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat. Genet. 2001, 27, 369–370. [Google Scholar] [CrossRef]
- Cerruti Mainardi, P.; Pastore, G.; Zweier, C.; Rauch, A. Mowat-Wilson syndrome and mutation in the zinc finger homeo box 1B gene: A well defined clinical entity. J. Med. Genet. 2004, 41, e16. [Google Scholar] [CrossRef][Green Version]
- Garavelli, L.; Mainardi, P.C. Mowat-Wilson syndrome. Orphanet J. Rare Dis. 2007, 2, 42. [Google Scholar] [CrossRef]
- Ishihara, N.; Yamada, K.; Yamada, Y.; Miura, K.; Kato, J.; Kuwabara, N.; Hara, Y.; Kobayashi, Y.; Hoshino, K.; Nomura, Y.; et al. Clinical and molecular analysis of Mowat-Wilson syndrome associated with ZFHX1B mutations and deletions at 2q22–q24.1. J. Med. Genet. 2004, 41, 387–393. [Google Scholar] [CrossRef][Green Version]
- Mowat, D.R.; Wilson, M.J.; Goossens, M. Mowat-Wilson syndrome. J. Med. Genet. 2003, 40, 305–310. [Google Scholar] [CrossRef]
- Yamada, K.; Yamada, Y.; Nomura, N.; Miura, K.; Wakako, R.; Hayakawa, C.; Matsumoto, A.; Kumagai, T.; Yoshimura, I.; Miyazaki, S.; et al. Nonsense and frameshift mutations in ZFHX1B, encoding Smad-interacting protein 1, cause a complex developmental disorder with a great variety of clinical features. Am. J. Hum. Genet. 2001, 69, 1178–1185. [Google Scholar] [CrossRef]
- Zweier, C.; Thiel, C.T.; Dufke, A.; Crow, Y.J.; Meinecke, P.; Suri, M.; Ala-Mello, S.; Beemer, F.; Bernasconi, S.; Bianchi, P.; et al. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur. J. Med. Genet. 2005, 48, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Zweier, C.; Horn, D.; Kraus, C.; Rauch, A. Atypical ZFHX1B mutation associated with a mild Mowat-Wilson syndrome phenotype. Am. J. Med. Genet. A 2006, 140, 869–872. [Google Scholar] [CrossRef]
- Garavelli, L.; Ivanovski, I.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; Baldo, C.; et al. Neuroimaging findings in Mowat-Wilson syndrome: A study of 54 patients. Genet. Med. 2017, 19, 691–700. [Google Scholar] [CrossRef]
- Ivanovski, I.; Djuric, O.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Rosato, S.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; et al. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet. Med. 2018, 20, 965–975. [Google Scholar] [CrossRef]
- Garavelli, L.; Donadio, A.; Zanacca, C.; Banchini, G.; Della Giustina, E.; Bertani, G.; Albertini, G.; Del Rossi, C.; Zweier, C.; Rauch, A.; et al. Hirschsprung disease, mental retardation, characteristic facial features, and mutation in the gene ZFHX1B (SIP1): Confirmation of the Mowat-Wilson syndrome. Am. J. Med. Genet. A 2003, 116a, 385–388. [Google Scholar] [CrossRef]
- Garavelli, L.; Zollino, M.; Mainardi, P.C.; Gurrieri, F.; Rivieri, F.; Soli, F.; Verri, R.; Albertini, E.; Favaron, E.; Zignani, M.; et al. Mowat-Wilson syndrome: Facial phenotype changing with age: Study of 19 Italian patients and review of the literature. Am. J. Med. Genet. A 2009, 149a, 417–426. [Google Scholar] [CrossRef]
- Zweier, C.; Albrecht, B.; Mitulla, B.; Behrens, R.; Beese, M.; Gillessen-Kaesbach, G.; Rott, H.D.; Rauch, A. “Mowat-Wilson” syndrome with and without Hirschsprung disease is a distinct, recognizable multiple congenital anomalies-mental retardation syndrome caused by mutations in the zinc finger homeo box 1B gene. Am. J. Med. Genet. 2002, 108, 177–181. [Google Scholar] [CrossRef]
- Wilson, M.; Mowat, D.; Dastot-Le Moal, F.; Cacheux, V.; Kääriäinen, H.; Cass, D.; Donnai, D.; Clayton-Smith, J.; Townshend, S.; Curry, C.; et al. Further delineation of the phenotype associated with heterozygous mutations in ZFHX1B. Am. J. Med. Genet. A 2003, 119a, 257–265. [Google Scholar] [CrossRef]
- Dastot-Le Moal, F.; Wilson, M.; Mowat, D.; Collot, N.; Niel, F.; Goossens, M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum. Mutat. 2007, 28, 313–321. [Google Scholar] [CrossRef]
- Heinritz, W.; Zweier, C.; Froster, U.G.; Strenge, S.; Kujat, A.; Syrbe, S.; Rauch, A.; Schuster, V. A missense mutation in the ZFHX1B gene associated with an atypical Mowat-Wilson syndrome phenotype. Am. J. Med. Genet. A 2006, 140, 1223–1227. [Google Scholar] [CrossRef]
- Ghoumid, J.; Drevillon, L.; Alavi-Naini, S.M.; Bondurand, N.; Rio, M.; Briand-Suleau, A.; Nasser, M.; Goodwin, L.; Raymond, P.; Yanicostas, C.; et al. ZEB2 zinc-finger missense mutations lead to hypomorphic alleles and a mild Mowat-Wilson syndrome. Hum. Mol. Genet. 2013, 22, 2652–2661. [Google Scholar] [CrossRef]
- Chng, Z.; Teo, A.; Pedersen, R.A.; Vallier, L. SIP1 mediates cell-fate decisions between neuroectoderm and mesendoderm in human pluripotent stem cells. Cell Stem Cell 2010, 6, 59–70. [Google Scholar] [CrossRef]
- Deryckere, A.; Stappers, E.; Dries, R.; Peyre, E.; van den Berghe, V.; Conidi, A.; Zampeta, F.I.; Francis, A.; Bresseleers, M.; Stryjewska, A.; et al. Multifaceted actions of Zeb2 in postnatal neurogenesis from the ventricular-subventricular zone to the olfactory bulb. Development 2020, 147. [Google Scholar] [CrossRef]
- Stryjewska, A.; Dries, R.; Pieters, T.; Verstappen, G.; Conidi, A.; Coddens, K.; Francis, A.; Umans, L.; van IJcken, W.F.; Berx, G.; et al. Zeb2 Regulates Cell Fate at the Exit from Epiblast State in Mouse Embryonic Stem Cells. Stem Cells 2017, 35, 611–625. [Google Scholar] [CrossRef]
- Goossens, S.; Janzen, V.; Bartunkova, S.; Yokomizo, T.; Drogat, B.; Crisan, M.; Haigh, K.; Seuntjens, E.; Umans, L.; Riedt, T.; et al. The EMT regulator Zeb2/Sip1 is essential for murine embryonic hematopoietic stem/progenitor cell differentiation and mobilization. Blood 2011, 117, 5620–5630. [Google Scholar] [CrossRef]
- Li, J.; Riedt, T.; Goossens, S.; Carrillo García, C.; Szczepanski, S.; Brandes, M.; Pieters, T.; Dobrosch, L.; Gütgemann, I.; Farla, N.; et al. The EMT transcription factor Zeb2 controls adult murine hematopoietic differentiation by regulating cytokine signaling. Blood 2017, 129, 460–472. [Google Scholar] [CrossRef]
- Boland, B.S.; He, Z.; Tsai, M.S.; Olvera, J.G.; Omilusik, K.D.; Duong, H.G.; Kim, E.S.; Limary, A.E.; Jin, W.; Milner, J.J.; et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Omilusik, K.D.; Best, J.A.; Yu, B.; Goossens, S.; Weidemann, A.; Nguyen, J.V.; Seuntjens, E.; Stryjewska, A.; Zweier, C.; Roychoudhuri, R.; et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med. 2015, 212, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Omilusik, K.D. ZEBs: Novel Players in Immune Cell Development and Function. Trends Immunol. 2019, 40, 431–446. [Google Scholar] [CrossRef]
- Scott, C.L.; Soen, B.; Martens, L.; Skrypek, N.; Saelens, W.; Taminau, J.; Blancke, G.; Van Isterdael, G.; Huylebroeck, D.; Haigh, J.; et al. The transcription factor Zeb2 regulates development of conventional and plasmacytoid DCs by repressing Id2. J. Exp. Med. 2016, 213, 897–911. [Google Scholar] [CrossRef]
- Scott, C.L.; T’Jonck, W.; Martens, L.; Todorov, H.; Sichien, D.; Soen, B.; Bonnardel, J.; De Prijck, S.; Vandamme, N.; Cannoodt, R.; et al. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity 2018, 49, 312–325.e5. [Google Scholar] [CrossRef]
- Van Helden, M.J.; Goossens, S.; Daussy, C.; Mathieu, A.L.; Faure, F.; Marçais, A.; Vandamme, N.; Farla, N.; Mayol, K.; Viel, S.; et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J. Exp. Med. 2015, 212, 2015–2025. [Google Scholar] [CrossRef]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef]
- Vandamme, N.; Denecker, G.; Bruneel, K.; Blancke, G.; Akay, Ö.; Taminau, J.; De Coninck, J.; De Smedt, E.; Skrypek, N.; Van Loocke, W.; et al. The EMT Transcription Factor ZEB2 Promotes Proliferation of Primary and Metastatic Melanoma while Suppressing an Invasive, Mesenchymal-Like Phenotype. Cancer Res. 2020, 80, 2983–2995. [Google Scholar] [CrossRef]
- Teraishi, M.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Higashi, Y.; Shimoda, S.; Asada, Y.; Hijikata, A.; Ohara, O.; Hiraki, Y.; et al. Critical involvement of ZEB2 in collagen fibrillogenesis: The molecular similarity between Mowat-Wilson syndrome and Ehlers-Danlos syndrome. Sci. Rep. 2017, 7, 46565. [Google Scholar] [CrossRef]
- Tatari, M.N.; De Craene, B.; Soen, B.; Taminau, J.; Vermassen, P.; Goossens, S.; Haigh, K.; Cazzola, S.; Lambert, J.; Huylebroeck, D.; et al. ZEB2-transgene expression in the epidermis compromises the integrity of the epidermal barrier through the repression of different tight junction proteins. Cell. Mol. Life Sci. 2014, 71, 3599–3609. [Google Scholar] [CrossRef]
- Rasouly, H.M.; Kumar, S.; Chan, S.; Pisarek-Horowitz, A.; Sharma, R.; Xi, Q.J.; Nishizaki, Y.; Higashi, Y.; Salant, D.J.; Maas, R.L.; et al. Loss of Zeb2 in mesenchyme-derived nephrons causes primary glomerulocystic disease. Kidney Int. 2016, 90, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Iwafuchi-Doi, M.; Matsuda, K.; Murakami, K.; Niwa, H.; Tesar, P.J.; Aruga, J.; Matsuo, I.; Kondoh, H. Transcriptional regulatory networks in epiblast cells and during anterior neural plate development as modeled in epiblast stem cells. Development 2012, 139, 3926–3937. [Google Scholar] [CrossRef] [PubMed]
- Seuntjens, E.; Nityanandam, A.; Miquelajauregui, A.; Debruyn, J.; Stryjewska, A.; Goebbels, S.; Nave, K.A.; Huylebroeck, D.; Tarabykin, V. Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat. Neurosci. 2009, 12, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Van de Putte, T.; Maruhashi, M.; Francis, A.; Nelles, L.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am. J. Hum. Genet. 2003, 72, 465–470. [Google Scholar] [CrossRef]
- Van Grunsven, L.A.; Papin, C.; Avalosse, B.; Opdecamp, K.; Huylebroeck, D.; Smith, J.C.; Bellefroid, E.J. XSIP1, a Xenopus zinc finger/homeodomain encoding gene highly expressed during early neural development. Mech. Dev. 2000, 94, 189–193. [Google Scholar] [CrossRef]
- Papin, C.; van Grunsven, L.A.; Verschueren, K.; Huylebroeck, D.; Smith, J.C. Dynamic regulation of Brachyury expression in the amphibian embryo by XSIP1. Mech. Dev. 2002, 111, 37–46. [Google Scholar] [CrossRef]
- van Grunsven, L.A.; Taelman, V.; Michiels, C.; Opdecamp, K.; Huylebroeck, D.; Bellefroid, E.J. deltaEF1 and SIP1 are differentially expressed and have overlapping activities during Xenopus embryogenesis. Dev. Dyn. 2006, 235, 1491–1500. [Google Scholar] [CrossRef]
- van Grunsven, L.A.; Taelman, V.; Michiels, C.; Verstappen, G.; Souopgui, J.; Nichane, M.; Moens, E.; Opdecamp, K.; Vanhomwegen, J.; Kricha, S.; et al. XSip1 neuralizing activity involves the co-repressor CtBP and occurs through BMP dependent and independent mechanisms. Dev. Biol. 2007, 306, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Maruhashi, M.; Nelles, L.; Van de Putte, T.; Verschueren, K.; Miyoshi, T.; Yoshimoto, A.; Kondoh, H.; Huylebroeck, D. Generation of the floxed allele of the SIP1 (Smad-interacting protein 1) gene for Cre-mediated conditional knockout in the mouse. Genesis 2002, 32, 82–84. [Google Scholar] [CrossRef]
- Delalande, J.M.; Guyote, M.E.; Smith, C.M.; Shepherd, I.T. Zebrafish sip1a and sip1b are essential for normal axial and neural patterning. Dev. Dyn. 2008, 237, 1060–1069. [Google Scholar] [CrossRef]
- Haenebalcke, L.; Goossens, S.; Naessens, M.; Kruse, N.; Farhang Ghahremani, M.; Bartunkova, S.; Haigh, K.; Pieters, T.; Dierickx, P.; Drogat, B.; et al. Efficient ROSA26-based conditional and/or inducible transgenesis using RMCE-compatible F1 hybrid mouse embryonic stem cells. Stem Cell Rev. Rep. 2013, 9, 774–785. [Google Scholar] [CrossRef]
- Kok, F.O.; Shepherd, I.T.; Sirotkin, H.I. Churchill and Sip1a repress fibroblast growth factor signaling during zebrafish somitogenesis. Dev. Dyn. 2010, 239, 548–558. [Google Scholar] [CrossRef]
- Maruhashi, M.; Van De Putte, T.; Huylebroeck, D.; Kondoh, H.; Higashi, Y. Involvement of SIP1 in positioning of somite boundaries in the mouse embryo. Dev. Dyn. 2005, 234, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, T.; Maruhashi, M.; Van De Putte, T.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Complementary expression pattern of Zfhx1 genes Sip1 and deltaEF1 in the mouse embryo and their genetic interaction revealed by compound mutants. Dev. Dyn. 2006, 235, 1941–1952. [Google Scholar] [CrossRef]
- Stanchina, L.; Van de Putte, T.; Goossens, M.; Huylebroeck, D.; Bondurand, N. Genetic interaction between Sox10 and Zfhx1b during enteric nervous system development. Dev. Biol. 2010, 341, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Stanchina, L.; Lecerf, L.; Gacem, N.; Conidi, A.; Baral, V.; Pingault, V.; Huylebroeck, D.; Bondurand, N. Differentiation of Mouse Enteric Nervous System Progenitor Cells Is Controlled by Endothelin 3 and Requires Regulation of Ednrb by SOX10 and ZEB2. Gastroenterology 2017, 152, 1139–1150.e4. [Google Scholar] [CrossRef] [PubMed]
- Miquelajauregui, A.; Van de Putte, T.; Polyakov, A.; Nityanandam, A.; Boppana, S.; Seuntjens, E.; Karabinos, A.; Higashi, Y.; Huylebroeck, D.; Tarabykin, V. Smad-interacting protein-1 (Zfhx1b) acts upstream of Wnt signaling in the mouse hippocampus and controls its formation. Proc. Natl. Acad. Sci. USA 2007, 104, 12919–12924. [Google Scholar] [CrossRef] [PubMed]
- Weng, Q.; Chen, Y.; Wang, H.; Xu, X.; Yang, B.; He, Q.; Shou, W.; Chen, Y.; Higashi, Y.; van den Berghe, V.; et al. Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 2012, 73, 713–728. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, G.L.; Lindtner, S.; Trzcinski, B.; Visel, A.; Pennacchio, L.A.; Huylebroeck, D.; Higashi, Y.; Rubenstein, J.L. Dlx1&2-dependent expression of Zfhx1b (Sip1, Zeb2) regulates the fate switch between cortical and striatal interneurons. Neuron 2013, 77, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, V.; Stappers, E.; Vandesande, B.; Dimidschstein, J.; Kroes, R.; Francis, A.; Conidi, A.; Lesage, F.; Dries, R.; Cazzola, S.; et al. Directed migration of cortical interneurons depends on the cell-autonomous action of Sip1. Neuron 2013, 77, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Nishizaki, Y.; Matsui, F.; Wakamatsu, N.; Higashi, Y. De novo inbred heterozygous Zeb2/Sip1 mutant mice uniquely generated by germ-line conditional knockout exhibit craniofacial, callosal and behavioral defects associated with Mowat-Wilson syndrome. Hum. Mol. Genet. 2015, 24, 6390–6402. [Google Scholar] [CrossRef]
- Srivatsa, S.; Parthasarathy, S.; Molnár, Z.; Tarabykin, V. Sip1 downstream Effector ninein controls neocortical axonal growth, ipsilateral branching, and microtubule growth and stability. Neuron 2015, 85, 998–1012. [Google Scholar] [CrossRef]
- Quintes, S.; Brinkmann, B.G.; Ebert, M.; Fröb, F.; Kungl, T.; Arlt, F.A.; Tarabykin, V.; Huylebroeck, D.; Meijer, D.; Suter, U.; et al. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat. Neurosci. 2016, 19, 1050–1059. [Google Scholar] [CrossRef]
- Wu, L.M.; Wang, J.; Conidi, A.; Zhao, C.; Wang, H.; Ford, Z.; Zhang, L.; Zweier, C.; Ayee, B.G.; Maurel, P.; et al. Zeb2 recruits HDAC-NuRD to inhibit Notch and controls Schwann cell differentiation and remyelination. Nat. Neurosci. 2016, 19, 1060–1072. [Google Scholar] [CrossRef]
- Hegarty, S.V.; Wyatt, S.L.; Howard, L.; Stappers, E.; Huylebroeck, D.; Sullivan, A.M.; O’Keeffe, G.W. Zeb2 is a negative regulator of midbrain dopaminergic axon growth and target innervation. Sci. Rep. 2017, 7, 8568. [Google Scholar] [CrossRef]
- Vivinetto, A.L.; Kim, I.D.; Goldberg, D.C.; Fones, L.; Brown, E.; Tarabykin, V.S.; Hill, C.E.; Cho, S.; Cave, J.W. Zeb2 Is a Regulator of Astrogliosis and Functional Recovery after CNS Injury. Cell Rep. 2020, 31, 107834. [Google Scholar] [CrossRef] [PubMed]
- Turovskaya, M.V.; Epifanova, E.A.; Tarabykin, V.S.; Babaev, A.A.; Turovsky, E.A. Interleukin-10 restores glutamate receptor-mediated Ca(2+)-signaling in brain circuits under loss of Sip1 transcription factor. Int. J. Neurosci. 2020, 1–12. [Google Scholar] [CrossRef]
- Wenger, T.L.; Harr, M.; Ricciardi, S.; Bhoj, E.; Santani, A.; Adam, M.P.; Barnett, S.S.; Ganetzky, R.; McDonald-McGinn, D.M.; Battaglia, D.; et al. “CHARGE-like presentation, craniosynostosis and mild Mowat-Wilson Syndrome diagnosed by recognition of the distinctive facial gestalt in a cohort of 28 new cases” American Journal of Medical Genetics Part A. 164:2557–2566, 2014. Am. J. Med. Genet. A 2015, 167, 1682–1683. [Google Scholar] [CrossRef]
- Jeub, M.; Emrich, M.; Pradier, B.; Taha, O.; Gailus-Durner, V.; Fuchs, H.; de Angelis, M.H.; Huylebroeck, D.; Zimmer, A.; Beck, H.; et al. The transcription factor Smad-interacting protein 1 controls pain sensitivity via modulation of DRG neuron excitability. Pain 2011, 152, 2384–2398. [Google Scholar] [CrossRef] [PubMed]
- Pradier, B.; Jeub, M.; Markert, A.; Mauer, D.; Tolksdorf, K.; Van de Putte, T.; Seuntjens, E.; Gailus-Durner, V.; Fuchs, H.; Hrabě de Angelis, M.; et al. Smad-interacting protein 1 affects acute and tonic, but not chronic pain. Eur. J. Pain 2014, 18, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Francius, C.; Rousso, D.L.; Seuntjens, E.; Debruyn, J.; Luxenhofer, G.; Huber, A.B.; Huylebroeck, D.; Novitch, B.G.; Clotman, F. Onecut transcription factors act upstream of Isl1 to regulate spinal motoneuron diversification. Development 2012, 139, 3109–3119. [Google Scholar] [CrossRef] [PubMed]
- Denecker, G.; Vandamme, N.; Akay, O.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef]
- Yoshimoto, A.; Saigou, Y.; Higashi, Y.; Kondoh, H. Regulation of ocular lens development by Smad-interacting protein 1 involving Foxe3 activation. Development 2005, 132, 4437–4448. [Google Scholar] [CrossRef] [PubMed]
- Manthey, A.L.; Lachke, S.A.; FitzGerald, P.G.; Mason, R.W.; Scheiblin, D.A.; McDonald, J.H.; Duncan, M.K. Loss of Sip1 leads to migration defects and retention of ectodermal markers during lens development. Mech. Dev. 2014, 131, 86–110. [Google Scholar] [CrossRef]
- Menuchin-Lasowski, Y.; Oren-Giladi, P.; Xie, Q.; Ezra-Elia, R.; Ofri, R.; Peled-Hajaj, S.; Farhy, C.; Higashi, Y.; Van de Putte, T.; Kondoh, H.; et al. Sip1 regulates the generation of the inner nuclear layer retinal cell lineages in mammals. Development 2016, 143, 2829–2841. [Google Scholar] [CrossRef]
- Wei, W.; Liu, B.; Jiang, H.; Jin, K.; Xiang, M. Requirement of the Mowat-Wilson Syndrome Gene Zeb2 in the Differentiation and Maintenance of Non-photoreceptor Cell Types During Retinal Development. Mol. Neurobiol. 2019, 56, 1719–1736. [Google Scholar] [CrossRef] [PubMed]
- Gladka, M.M.; Kohela, A.; Molenaar, B.; Versteeg, D.; Kooijman, L.; Monshouwer-Kloots, J.; Kremer, V.; Vos, H.R.; Huibers, M.M.H.; Haigh, J.J.; et al. Cardiomyocytes stimulate angiogenesis after ischemic injury in a ZEB2-dependent manner. Nat. Commun. 2021, 12, 84. [Google Scholar] [CrossRef]
- De Haan, W.; Dheedene, W.; Apelt, K.; Décombas-Deschamps, S.; Vinckier, S.; Verhulst, S.; Conidi, A.; Deffieux, T.; Staring, M.W.; Vandervoort, P.; et al. Endothelial Zeb2 preserves the hepatic angioarchitecture and protects against liver fibrosis. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bruneel, K.; Verstappe, J.; Vandamme, N.; Berx, G. Intrinsic Balance between ZEB Family Members Is Important for Melanocyte Homeostasis and Melanoma Progression. Cancers 2020, 12, 2248. [Google Scholar] [CrossRef]
- Conidi, A.; Cazzola, S.; Beets, K.; Coddens, K.; Collart, C.; Cornelis, F.; Cox, L.; Joke, D.; Dobreva, M.P.; Dries, R.; et al. Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGFβ/BMP signaling in vivo. Cytokine Growth Factor Rev. 2011, 22, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. Zeb2: A multifunctional regulator of nervous system development. Prog. Neurobiol. 2015, 132, 81–95. [Google Scholar] [CrossRef]
- Menuchin-Lasowski, Y.; Dagan, B.; Conidi, A.; Cohen-Gulkar, M.; David, A.; Ehrlich, M.; Giladi, P.O.; Clark, B.S.; Blackshaw, S.; Shapira, K.; et al. Zeb2 regulates the balance between retinal interneurons and Müller glia by inhibition of BMP-Smad signaling. Dev. Biol. 2020, 468, 80–92. [Google Scholar] [CrossRef]
- Yasumi, T.; Inoue, M.; Maruhashi, M.; Kamachi, Y.; Higashi, Y.; Kondoh, H.; Uchikawa, M. Regulation of trunk neural crest delamination by δEF1 and Sip1 in the chicken embryo. Dev. Growth Differ. 2016, 58, 205–214. [Google Scholar] [CrossRef][Green Version]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schujiers, J.; Lee, T.I.; Zhao, K.; et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef]
- Benito-Kwiecinski, S.; Giandomenico, S.L.; Sutcliffe, M.; Riis, E.S.; Freire-Pritchett, P.; Kelava, I.; Wunderlich, S.; Martin, U.; Wray, G.A.; McDole, K.; et al. An early cell shape transition drives evolutionary expansion of the human forebrain. Cell 2021, 184, 2084–2102.e19. [Google Scholar] [CrossRef]
- Nelles, L.; Van de Putte, T.; van Grunsven, L.; Huylebroeck, D.; Verschueren, K. Organization of the mouse Zfhx1b gene encoding the two-handed zinc finger repressor Smad-interacting protein-1. Genomics 2003, 82, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Conidi, A.; van den Berghe, V.; Leslie, K.; Stryjewska, A.; Xue, H.; Chen, Y.G.; Seuntjens, E.; Huylebroeck, D. Four amino acids within a tandem QxVx repeat in a predicted extended α-helix of the Smad-binding domain of Sip1 are necessary for binding to activated Smad proteins. PLoS ONE 2013, 8, e76733. [Google Scholar] [CrossRef]
- Van Grunsven, L.A.; Huylebroeck, D.; Verschueren, K. Complex Smad-dependent transcriptional responses in vertebrate development and human disease. Crit. Rev. Eukaryot. Gene Expr. 2002, 12, 101–118. [Google Scholar] [CrossRef]
- Higashi, Y.; Moribe, H.; Takagi, T.; Sekido, R.; Kawakami, K.; Kikutani, H.; Kondoh, H. Impairment of T cell development in deltaEF1 mutant mice. J. Exp. Med. 1997, 185, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Moribe, H.; Kondoh, H.; Higashi, Y. DeltaEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development 1998, 125, 21–31. [Google Scholar] [CrossRef]
- Remacle, J.E.; Kraft, H.; Lerchner, W.; Wuytens, G.; Collart, C.; Verschueren, K.; Smith, J.C.; Huylebroeck, D. New mode of DNA binding of multi-zinc finger transcription factors: DeltaEF1 family members bind with two hands to two target sites. EMBO J. 1999, 18, 5073–5084. [Google Scholar] [CrossRef]
- Moribe, H.; Takagi, T.; Kondoh, H.; Higashi, Y. Suppression of polydactyly of the Gli3 mutant (extra toes) by deltaEF1 homozygous mutation. Dev. Growth Differ. 2000, 42, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Sekido, R.; Takagi, T.; Okanami, M.; Moribe, H.; Yamamura, M.; Higashi, Y.; Kondoh, H. Organization of the gene encoding transcriptional repressor deltaEF1 and cross-species conservation of its domains. Gene 1996, 173, 227–232. [Google Scholar] [CrossRef]
- Van Grunsven, L.A.; Schellens, A.; Huylebroeck, D.; Verschueren, K. SIP1 (Smad interacting protein 1) and deltaEF1 (delta-crystallin enhancer binding factor) are structurally similar transcriptional repressors. J. Bone Joint Surg. Am. 2001, 83, S40–S47. [Google Scholar] [CrossRef]
- Sekido, R.; Murai, K.; Funahashi, J.; Kamachi, Y.; Fujisawa-Sehara, A.; Nabeshima, Y.; Kondoh, H. The delta-crystallin enhancer-binding protein delta EF1 is a repressor of E2-box-mediated gene activation. Mol. Cell. Biol. 1994, 14, 5692–5700. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.; van Grunsven, L.A.; Michiels, C.; Van de Putte, T.; Souopgui, J.; Van Damme, J.; Bellefroid, E.; Vandekerckhove, J.; Huylebroeck, D. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum. Mol. Genet. 2008, 17, 1175–1183. [Google Scholar] [CrossRef]
- Chinnadurai, G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell 2002, 9, 213–224. [Google Scholar] [CrossRef]
- Chinnadurai, G. CtBP family proteins: More than transcriptional corepressors. Bioessays 2003, 25, 9–12. [Google Scholar] [CrossRef]
- Zhao, L.J.; Kuppuswamy, M.; Vijayalingam, S.; Chinnadurai, G. Interaction of ZEB and histone deacetylase with the PLDLS-binding cleft region of monomeric C-terminal binding protein 2. BMC Mol. Biol. 2009, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Subramanian, T.; Vijayalingam, S.; Chinnadurai, G. PLDLS-dependent interaction of E1A with CtBP: Regulation of CtBP nuclear localization and transcriptional functions. Oncogene 2007, 26, 7544–7551. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G. Transcriptional regulation by C-terminal binding proteins. Int. J. Biochem. Cell Biol. 2007, 39, 1593–1607. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sawada, J.; Sui, G.; Affarel, B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef]
- Stankiewicz, T.R.; Gray, J.J.; Winter, A.N.; Linseman, D.A. C-terminal binding proteins: Central players in development and disease. Biomol. Concepts 2014, 5, 489–511. [Google Scholar] [CrossRef]
- Subramanian, T.; Chinnadurai, G. Association of class I histone deacetylases with transcriptional corepressor CtBP. FEBS Lett. 2003, 540, 255–258. [Google Scholar] [CrossRef]
- Long, J.; Zuo, D.; Park, M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef]
- Van Grunsven, L.A.; Michiels, C.; Van de Putte, T.; Nelles, L.; Wuytens, G.; Verschueren, K.; Huylebroeck, D. Interaction between Smad-interacting protein-1 and the corepressor C-terminal binding protein is dispensable for transcriptional repression of E-cadherin. J. Biol. Chem. 2003, 278, 26135–26145. [Google Scholar] [CrossRef]
- Liu, S.; Long, J.; Yuan, B.; Zheng, M.; Xiao, M.; Xu, J.; Lin, X.; Feng, X.H. SUMO Modification Reverses Inhibitory Effects of Smad Nuclear Interacting Protein-1 in TGF-β Responses. J. Biol. Chem. 2016, 291, 24418–24430. [Google Scholar] [CrossRef]
- Fujii, M.; Lyakh, L.A.; Bracken, C.P.; Fukuoka, J.; Hayakawa, M.; Tsukiyama, T.; Soll, S.J.; Harris, M.; Rocha, S.; Roche, K.C.; et al. SNIP1 is a candidate modifier of the transcriptional activity of c-Myc on E box-dependent target genes. Mol. Cell 2006, 24, 771–783. [Google Scholar] [CrossRef]
- Bassez, G.; Camand, O.J.; Cacheux, V.; Kobetz, A.; Dastot-Le Moal, F.; Marchant, D.; Catala, M.; Abitbol, M.; Goossens, M. Pleiotropic and diverse expression of ZFHX1B gene transcripts during mouse and human development supports the various clinical manifestations of the “Mowat-Wilson” syndrome. Neurobiol. Dis. 2004, 15, 240–250. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Alvarez, A.B.; Peña, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Macho-Maschler, S.; Custic, I.; Niemiec, J.; Guío-Carrión, A.; Hasenfuss, S.C.; Eger, A.; Müller, M.; Beug, H.; Wagner, E.F. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ. 2015, 22, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Integrative genomic analyses of ZEB2: Transcriptional regulation of ZEB2 based on SMADs, ETS1, HIF1alpha, POU/OCT, and NF-kappaB. Int. J. Oncol. 2009, 34, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Sinh, N.D.; Endo, K.; Miyazawa, K.; Saitoh, M. Ets1 and ESE1 reciprocally regulate expression of ZEB1/ZEB2, dependent on ERK1/2 activity, in breast cancer cells. Cancer Sci. 2017, 108, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, X.; Qiao, W.; Kong, L.; Sun, D.; Li, Z. Transcription factor E2F1 promotes EMT by regulating ZEB2 in small cell lung cancer. BMC Cancer 2017, 17, 719. [Google Scholar] [CrossRef]
- Xia, L.; Huang, W.; Tian, D.; Zhang, L.; Qi, X.; Chen, Z.; Shang, X.; Nie, Y.; Wu, K. Forkhead box Q1 promotes hepatocellular carcinoma metastasis by transactivating ZEB2 and VersicanV1 expression. Hepatology 2014, 59, 958–973. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, C.; Gao, W.; Chen, T.; Qian, T.; Hu, J.; Tan, Y. FOXA2 attenuates the epithelial to mesenchymal transition by regulating the transcription of E-cadherin and ZEB2 in human breast cancer. Cancer Lett. 2015, 361, 240–250. [Google Scholar] [CrossRef] [PubMed]
- El-Kasti, M.M.; Wells, T.; Carter, D.A. A novel long-range enhancer regulates postnatal expression of Zeb2: Implications for Mowat-Wilson syndrome phenotypes. Hum. Mol. Genet. 2012, 21, 5429–5442. [Google Scholar] [CrossRef] [PubMed]
- Bar Yaacov, R.; Eshel, R.; Farhi, E.; Shemuluvich, F.; Kaplan, T.; Birnbaum, R.Y. Functional characterization of the ZEB2 regulatory landscape. Hum. Mol. Genet. 2019, 28, 1487–1497. [Google Scholar] [CrossRef]
- Birkhoff, J.C.; Brouwer, R.W.W.; Kolovos, P.; Korporaal, A.L.; Bermejo-Santos, A.; Boltsis, I.; Nowosad, K.; van den Hout, M.; Grosveld, F.G.; van IJcken, W.F.J.; et al. Targeted chromatin conformation analysis identifies novel distal neural enhancers of ZEB2 in pluripotent stem cell differentiation. Hum. Mol. Genet. 2020, 29, 2535–2550. [Google Scholar] [CrossRef] [PubMed]
- Beclin, C.; Follert, P.; Stappers, E.; Barral, S.; Coré, N.; de Chevigny, A.; Magnone, V.; Lebrigand, K.; Bissels, U.; Huylebroeck, D.; et al. miR-200 family controls late steps of postnatal forebrain neurogenesis via Zeb2 inhibition. Sci. Rep. 2016, 6, 35729. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, N.R.; Silahtaroglu, A.; Orom, U.A.; Kauppinen, S.; Lund, A.H. miR-200b mediates post-transcriptional repression of ZFHX1B. RNA 2007, 13, 1172–1178. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef]
- Perdigão-Henriques, R.; Petrocca, F.; Altschuler, G.; Thomas, M.P.; Le, M.T.; Tan, S.M.; Hide, W.; Lieberman, J. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene 2016, 35, 158–172. [Google Scholar] [CrossRef]
- Wang, G.; Guo, X.; Hong, W.; Liu, Q.; Wei, T.; Lu, C.; Gao, L.; Ye, D.; Zhou, Y.; Chen, J.; et al. Critical regulation of miR-200/ZEB2 pathway in Oct4/Sox2-induced mesenchymal-to-epithelial transition and induced pluripotent stem cell generation. Proc. Natl. Acad. Sci. USA 2013, 110, 2858–2863. [Google Scholar] [CrossRef]
- Bracken, C.P.; Gregory, P.A.; Khew-Goodall, Y.; Goodall, G.J. The role of microRNAs in metastasis and epithelial-mesenchymal transition. Cell. Mol. Life Sci. 2009, 66, 1682–1699. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Kropivšek, K.; Pickford, J.; Carter, D.A. Postnatal dynamics of Zeb2 expression in rat brain: Analysis of novel 3′ UTR sequence reveals a miR-9 interacting site. J. Mol. Neurosci. 2014, 52, 138–147. [Google Scholar] [CrossRef]
- Kato, M.; Zhang, J.; Wang, M.; Lanting, L.; Yuan, H.; Rossi, J.J.; Natarajan, R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. USA 2007, 104, 3432–3437. [Google Scholar] [CrossRef]
- Yang, L.P.; Lin, Q.; Mu, X.L. MicroRNA-155 and FOXP3 jointly inhibit the migration and invasion of colorectal cancer cells by regulating ZEB2 expression. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6131–6138. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lu, Y.; Yang, P.; Chen, Q.; Wang, Y.; Ding, Q.; Xu, T.; Li, X.; Li, C.; Huang, C.; et al. MicroRNA-145 induces the senescence of activated hepatic stellate cells through the activation of p53 pathway by ZEB2. J. Cell. Physiol. 2019, 234, 7587–7599. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Wang, M.; Guo, W.; Huang, S.; Wang, Z.; Zhao, X.; Du, H.; Song, L.; Peng, X. Double-negative feedback loop between ZEB2 and miR-145 regulates epithelial-mesenchymal transition and stem cell properties in prostate cancer cells. Cell Tissue Res. 2014, 358, 763–778. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tang, Z.Y.; He, Y.; Liu, L.F.; Li, D.J.; Chen, X. miRNA-205 is a candidate tumor suppressor that targets ZEB2 in renal cell carcinoma. Oncol. Res. Treat. 2014, 37, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Lu, Y.X.; Zhang, J.X.; Wei, X.L.; Wang, F.; Zeng, Z.L.; Pan, Z.Z.; Yuan, Y.F.; Wang, F.H.; Pelicano, H.; et al. Long non-coding RNA UICLM promotes colorectal cancer liver metastasis by acting as a ceRNA for microRNA-215 to regulate ZEB2 expression. Theranostics 2017, 7, 4836–4849. [Google Scholar] [CrossRef]
- Di Gennaro, A.; Damiano, V.; Brisotto, G.; Armellin, M.; Perin, T.; Zucchetto, A.; Guardascione, M.; Spaink, H.P.; Doglioni, C.; Snaar-Jagalska, B.E.; et al. A p53/miR-30a/ZEB2 axis controls triple negative breast cancer aggressiveness. Cell Death Differ. 2018, 25, 2165–2180. [Google Scholar] [CrossRef]
- Guan, T.; Dominguez, C.X.; Amezquita, R.A.; Laidlaw, B.J.; Cheng, J.; Henao-Mejia, J.; Williams, A.; Flavell, R.A.; Lu, J.; Kaech, S.M. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8+ T cell fates. J. Exp. Med. 2018, 215, 1153–1168. [Google Scholar] [CrossRef]
- Ren, Z.; Yang, T.; Ding, J.; Liu, W.; Meng, X.; Zhang, P.; Liu, K.; Wang, P. MiR-520d-3p antitumor activity in human breast cancer via post-transcriptional regulation of spindle and kinetochore associated 2 expression. Am. J. Transl. Res. 2018, 10, 1097–1108. [Google Scholar] [PubMed]
- Wang, L.; Wei, Z.; Wu, K.; Dai, W.; Zhang, C.; Peng, J.; He, Y. Long noncoding RNA B3GALT5-AS1 suppresses colon cancer liver metastasis via repressing microRNA-203. Aging 2018, 10, 3662–3682. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y. miR-146 promotes HBV replication and expression by targeting ZEB2. Biomed. Pharmacother. 2018, 99, 576–582. [Google Scholar] [CrossRef]
- Chen, X.F.; Guo, J.F.; Xu, J.F.; Yin, S.H.; Cao, W.L. MiRNA-206 inhibits proliferation of renal clear cell carcinoma by targeting ZEB2. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7826–7834. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Zhang, S.; Xu, W.; Shi, D.; Zhuo, M.; Liang, S.; Lei, W.; Xie, C. MicroRNA-30a regulates cell proliferation, migration, invasion and apoptosis in human nasopharyngeal carcinoma via targeted regulation of ZEB2. Mol. Med. Rep. 2019, 20, 1672–1682. [Google Scholar] [CrossRef]
- Feng, S.; Liu, W.; Bai, X.; Pan, W.; Jia, Z.; Zhang, S.; Zhu, Y.; Tan, W. Corrigendum to’LncRNA-CTS promotes metastasis and epithelial-to-mesenchymal transition through regulating miR-505/ZEB2 axis in cervical cancer’ [Cancer Lett. 465 (2019) 105–117]. Cancer Lett. 2020, 493, 178. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.B.; Gao, F.Z.; Chen, X.F. MiRNA-1179 suppresses the metastasis of hepatocellular carcinoma by interacting with ZEB2. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5149–5157. [Google Scholar] [CrossRef]
- Lin, H.; Zheng, X.; Lu, T.; Gu, Y.; Zheng, C.; Yan, H. The proliferation and invasion of osteosarcoma are inhibited by miR-101 via targetting ZEB2. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.X.; Shi, X.C.; Zai, L.Q.; Bi, H.; Yang, Q. LncRNA CASC19 promotes the proliferation, migration and invasion of non-small cell lung carcinoma via regulating miRNA-130b-3p. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 247–255. [Google Scholar] [CrossRef]
- Xu, R.; Zhou, F.; Yu, T.; Xu, G.; Zhang, J.; Wang, Y.; Zhao, L.; Liu, N. MicroRNA-940 inhibits epithelial-mesenchymal transition of glioma cells via targeting ZEB2. Am. J. Transl. Res. 2019, 11, 7351–7363. [Google Scholar] [PubMed]
- Zhang, X.; Xu, X.; Ge, G.; Zang, X.; Shao, M.; Zou, S.; Zhang, Y.; Mao, Z.; Zhang, J.; Mao, F.; et al. miR-498 inhibits the growth and metastasis of liver cancer by targeting ZEB2. Oncol. Rep. 2019, 41, 1638–1648. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Qin, Y.; Chen, C.; Yang, J.; Song, N.; Gu, M. MicroRNA-200c-3p/ZEB2 loop plays a crucial role in the tumor progression of prostate carcinoma. Ann. Transl. Med. 2019, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhong, Y.; Li, L. miR-124 and miR-203 synergistically inactivate EMT pathway via coregulation of ZEB2 in clear cell renal cell carcinoma (ccRCC). J. Transl. Med. 2020, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Wang, Y.; Zhao, L.; Zou, W.; Tan, M.; He, Q. Exosomal miRNA-215-5p Derived from Adipose-Derived Stem Cells Attenuates Epithelial-Mesenchymal Transition of Podocytes by Inhibiting ZEB2. Biomed. Res. Int. 2020, 2020, 2685305. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Yu, C.C.; Liao, Y.W.; Hsieh, P.L.; Chu, P.M.; Liu, C.M.; Yu, C.H.; Su, T.R. miR-200a inhibits proliferation rate in drug-induced gingival overgrowth through targeting ZEB2. J. Formos. Med. Assoc. 2020, 119, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, Y.; Li, W.; Han, C.; Zhou, L.; Hu, H. MicroRNA-200c-3p inhibits proliferation and migration of renal artery endothelial cells by directly targeting ZEB2. Exp. Cell Res. 2020, 387, 111778. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xuan, W.; Wang, H.; Sun, F.; Zhang, C.; Gong, Q.; Ge, S. miR-200b regulates cellular senescence and inflammatory responses by targeting ZEB2 in pulmonary emphysema. Artif. Cells Nanomed. Biotechnol. 2020, 48, 656–663. [Google Scholar] [CrossRef]
- Yan, Z.; Bi, M.; Zhang, Q.; Song, Y.; Hong, S. LncRNA TUG1 promotes the progression of colorectal cancer via the miR-138-5p/ZEB2 axis. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Shen, W.; Hu, H.; Li, C.; Han, G. MicroRNA-140 Represses Esophageal Cancer Progression via Targeting ZEB2 to Regulate Wnt/β-Catenin Pathway. J. Surg. Res. 2021, 257, 267–277. [Google Scholar] [CrossRef]
- Lerchner, W.; Latinkic, B.V.; Remacle, J.E.; Huylebroeck, D.; Smith, J.C. Region-specific activation of the Xenopus brachyury promoter involves active repression in ectoderm and endoderm: A study using transgenic frog embryos. Development 2000, 127, 2729–2739. [Google Scholar] [CrossRef]
- Conlon, F.L.; Smith, J.C. Interference with brachyury function inhibits convergent extension, causes apoptosis, and reveals separate requirements in the FGF and activin signalling pathways. Dev. Biol. 1999, 213, 85–100. [Google Scholar] [CrossRef]
- Nitta, K.R.; Takahashi, S.; Haramoto, Y.; Fukuda, M.; Tanegashima, K.; Onuma, Y.; Asashima, M. The N-terminus zinc finger domain of Xenopus SIP1 is important for neural induction, but not for suppression of Xbra expression. Int. J. Dev. Biol. 2007, 51, 321–325. [Google Scholar] [CrossRef]
- Nishizaki, Y.; Takagi, T.; Matsui, F.; Higashi, Y. SIP1 expression patterns in brain investigated by generating a SIP1-EGFP reporter knock-in mouse. Genesis 2014, 52, 56–67. [Google Scholar] [CrossRef]
- Pla, P.; Monsoro-Burq, A.H. The neural border: Induction, specification and maturation of the territory that generates neural crest cells. Dev. Biol. 2018, 444, S36–S46. [Google Scholar] [CrossRef] [PubMed]
- Seal, S.; Monsoro-Burq, A.H. Insights into the Early Gene Regulatory Network Controlling Neural Crest and Placode Fate Choices at the Neural Border. Front. Physiol. 2020, 11, 608812. [Google Scholar] [CrossRef] [PubMed]
- Shyamala, K.; Yanduri, S.; Girish, H.C.; Murgod, S. Neural crest: The fourth germ layer. J. Oral Maxillofac. Pathol. 2015, 19, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Bronner, M.E. Neural crest lineage analysis: From past to future trajectory. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Thawani, A.; Groves, A.K. Building the Border: Development of the Chordate Neural Plate Border Region and Its Derivatives. Front. Physiol. 2020, 11, 608880. [Google Scholar] [CrossRef] [PubMed]
- LaBonne, C.; Bronner-Fraser, M. Molecular mechanisms of neural crest formation. Annu. Rev. Cell Dev. Biol. 1999, 15, 81–112. [Google Scholar] [CrossRef]
- Stuhlmiller, T.J.; García-Castro, M.I. Current perspectives of the signaling pathways directing neural crest induction. Cell. Mol. Life Sci. 2012, 69, 3715–3737. [Google Scholar] [CrossRef]
- Shin, J.O.; Kim, E.J.; Cho, K.W.; Nakagawa, E.; Kwon, H.J.; Cho, S.W.; Jung, H.S. BMP4 signaling mediates Zeb family in developing mouse tooth. Histochem. Cell Biol. 2012, 137, 791–800. [Google Scholar] [CrossRef]
- Adam, M.P.; Schelley, S.; Gallagher, R.; Brady, A.N.; Barr, K.; Blumberg, B.; Shieh, J.T.; Graham, J.; Slavotinek, A.; Martin, M.; et al. Clinical features and management issues in Mowat-Wilson syndrome. Am. J. Med. Genet. A 2006, 140, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Hjerling-Leffler, J.; Marmigère, F.; Heglind, M.; Cederberg, A.; Koltzenburg, M.; Enerbäck, S.; Ernfors, P. The boundary cap: A source of neural crest stem cells that generate multiple sensory neuron subtypes. Development 2005, 132, 2623–2632. [Google Scholar] [CrossRef]
- Maro, G.S.; Vermeren, M.; Voiculescu, O.; Melton, L.; Cohen, J.; Charnay, P.; Topilko, P. Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nat. Neurosci. 2004, 7, 930–938. [Google Scholar] [CrossRef]
- Amiel, J.; Sproat-Emison, E.; Garcia-Barcelo, M.; Lantieri, F.; Burzynski, G.; Borrego, S.; Pelet, A.; Arnold, S.; Miao, X.; Griseri, P.; et al. Hirschsprung disease, associated syndromes and genetics: A review. J. Med. Genet. 2008, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marín, O.; Valiente, M.; Ge, X.; Tsai, L.H. Guiding neuronal cell migrations. Cold Spring Harb. Perspect. Biol. 2010, 2, a001834. [Google Scholar] [CrossRef] [PubMed]
- Levitt, P.; Eagleson, K.L.; Powell, E.M. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004, 27, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.M.; Campbell, D.B.; Stanwood, G.D.; Davis, C.; Noebels, J.L.; Levitt, P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J. Neurosci. 2003, 23, 622–631. [Google Scholar] [CrossRef]
- Cordelli, D.M.; Pellicciari, A.; Kiriazopulos, D.; Franzoni, E.; Garavelli, L. Epilepsy in Mowat-Wilson syndrome: Is it a matter of GABA? Epilepsia 2013, 54, 1331–1332. [Google Scholar] [CrossRef] [PubMed]
- Conces, M.R.; Hughes, A.; Pierson, C.R. Neuropathology of Mowat-Wilson Syndrome. Pediatr. Dev. Pathol. 2020, 23, 322–325. [Google Scholar] [CrossRef]
- Fuentealba, L.C.; Rompani, S.B.; Parraguez, J.I.; Obernier, K.; Romero, R.; Cepko, C.L.; Alvarez-Buylla, A. Embryonic Origin of Postnatal Neural Stem Cells. Cell 2015, 161, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Young, K.M.; Fogarty, M.; Kessaris, N.; Richardson, W.D. Subventricular zone stem cells are heterogeneous with respect to their embryonic origins and neurogenic fates in the adult olfactory bulb. J. Neurosci. 2007, 27, 8286–8296. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Alvarez-Buylla, A. The Adult Ventricular-Subventricular Zone (V-SVZ) and Olfactory Bulb (OB) Neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Buffo, A.; Rite, I.; Tripathi, P.; Lepier, A.; Colak, D.; Horn, A.P.; Mori, T.; Götz, M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc. Natl. Acad. Sci. USA 2008, 105, 3581–3586. [Google Scholar] [CrossRef]
- Pekny, M.; Wilhelmsson, U.; Tatlisumak, T.; Pekna, M. Astrocyte activation and reactive gliosis-A new target in stroke? Neurosci Lett. 2019, 689, 45–55. [Google Scholar] [CrossRef]
- Mattugini, N.; Bocchi, R.; Scheuss, V.; Russo, G.L.; Torper, O.; Lao, C.L.; Götz, M. Inducing Different Neuronal Subtypes from Astrocytes in the Injured Mouse Cerebral Cortex. Neuron 2019, 103, 1086–1095.e5. [Google Scholar] [CrossRef]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef]
- Okada, S.; Hara, M.; Kobayakawa, K.; Matsumoto, Y.; Nakashima, Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci. Res. 2018, 126, 39–43. [Google Scholar] [CrossRef]
- Madelaine, R.; Mourrain, P. Endogenous retinal neural stem cell reprogramming for neuronal regeneration. Neural Regen. Res. 2017, 12, 1765–1767. [Google Scholar] [CrossRef]
- McCallum, S.; Obata, Y.; Fourli, E.; Boeing, S.; Peddie, C.J.; Xu, Q.; Horswell, S.; Kelsh, R.N.; Collinson, L.; Wilkinson, D.; et al. Enteric glia as a source of neural progenitors in adult zebrafish. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Memic, F.; Knoflach, V.; Morarach, K.; Sadler, R.; Laranjeira, C.; Hjerling-Leffler, J.; Sundström, E.; Pachnis, V.; Marklund, U. Transcription and Signaling Regulators in Developing Neuronal Subtypes of Mouse and Human Enteric Nervous System. Gastroenterology 2018, 154, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, S.; Song, X.; Yuan, J.; Qi, D.; Gu, X.; Yin, M.Y.; Han, Z.; Zhu, Y.; Liu, Z.; et al. Glial Cell-Based Vascular Mechanisms and Transplantation Therapies in Brain Vessel and Neurodegenerative Diseases. Front. Cell. Neurosci. 2021, 15, 627682. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Toledo, E.M.; Rosmaninho, P.; Peng, C.; Uhlén, P.; Castro, D.S.; Arenas, E. A Zeb2-miR-200c loop controls midbrain dopaminergic neuron neurogenesis and migration. Commun. Biol. 2018, 1, 75. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Yu, K.; Lu, F.; Wang, J.; Wu, L.N.; Zhao, C.; Li, Q.; Zhou, X.; Liu, H.; Mu, D.; et al. Transcriptional Regulator ZEB2 Is Essential for Bergmann Glia Development. J. Neurosci. 2018, 38, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Khor, C.C.; Miyake, M.; Chen, L.J.; Shi, Y.; Barathi, V.A.; Qiao, F.; Nakata, I.; Yamashiro, K.; Zhou, X.; Tam, P.O.; et al. Genome-wide association study identifies ZFHX1B as a susceptibility locus for severe myopia. Hum. Mol. Genet. 2013, 22, 5288–5294. [Google Scholar] [CrossRef]
- Ariss, M.; Natan, K.; Friedman, N.; Traboulsi, E.I. Ophthalmologic abnormalities in Mowat-Wilson syndrome and a mutation in ZEB2. Ophthalmic Genet. 2012, 33, 159–160. [Google Scholar] [CrossRef]
- Gregory-Evans, C.Y.; Williams, M.J.; Halford, S.; Gregory-Evans, K. Ocular coloboma: A reassessment in the age of molecular neuroscience. J. Med. Genet. 2004, 41, 881–891. [Google Scholar] [CrossRef]
- McGaughran, J.; Sinnott, S.; Dastot-Le Moal, F.; Wilson, M.; Mowat, D.; Sutton, B.; Goossens, M. Recurrence of Mowat-Wilson syndrome in siblings with the same proven mutation. Am. J. Med. Genet. A 2005, 137a, 302–304. [Google Scholar] [CrossRef]
- Tanteles, G.A.; Christophidou-Anastasiadou, V. Ocular phenotype of Mowat-Wilson syndrome in the first reported Cypriot patients: An under-recognized association. Clin. Dysmorphol. 2014, 23, 20–23. [Google Scholar] [CrossRef]
- Eisaki, A.; Kuroda, H.; Fukui, A.; Asashima, M. XSIP1, a member of two-handed zinc finger proteins, induced anterior neural markers in Xenopus laevis animal cap. Biochem. Biophys. Res. Commun. 2000, 271, 151–157. [Google Scholar] [CrossRef]
- Farhy, C.; Elgart, M.; Shapira, Z.; Oron-Karni, V.; Yaron, O.; Menuchin, Y.; Rechavi, G.; Ashery-Padan, R. Pax6 is required for normal cell-cycle exit and the differentiation kinetics of retinal progenitor cells. PLoS ONE 2013, 8, e76489. [Google Scholar] [CrossRef]
- Zalc, B.; Fields, R.D. Do Action Potentials Regulate Myelination? Neuroscientist 2000, 6, 5–13. [Google Scholar] [CrossRef]
- Kotter, M.R.; Stadelmann, C.; Hartung, H.P. Enhancing remyelination in disease—Can we wrap it up? Brain 2011, 134, 1882–1900. [Google Scholar] [CrossRef]
- Di Filippo, E.S.; Costamagna, D.; Giacomazzi, G.; Cortés-Calabuig, Á.; Stryjewska, A.; Huylebroeck, D.; Fulle, S.; Sampaolesi, M. Zeb2 Regulates Myogenic Differentiation in Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 2525. [Google Scholar] [CrossRef]
- Schuster, J.; Sobol, M.; Fatima, A.; Khalfallah, A.; Laan, L.; Anderlid, B.M.; Nordgren, A.; Dahl, N. Mowat-Wilson syndrome: Generation of two human iPS cell lines (UUIGPi004A and UUIGPi005A) from siblings with a truncating ZEB2 gene variant. Stem Cell Res. 2019, 39, 101518. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Model | Phenotype Underlying MOWS-Like Defects | Publication |
|---|---|---|
| SIP1 flox(ex7) SIP1 flox(ex7) EIIa-Cre; Zeb2Δex7/Δex7 | Early post-gastrulation embryonic lethality, failure of neural tube closure, cranial NCC delamination and migration, and vagal NCC generation, defected somite boundary positioning | Higashi et al., 2002, van de Putte et al., 2003, Maruhashi et al., 2005 [48,53,57] |
| Zp3-Cre; Zeb2+/Δex7; ΔEF1+/− | Defect in somite production and developmental arrest at E8.5, severe defected dorsal neural tube | Miyoshi et al., 2006 [58] |
| Emx1-Cre+/−; Zeb2KO/Δex7 | Lack of hippocampus and corpus callosum | Miquelajauregui et al., 2007 [61] |
| Wnt1-Cre+/−; Zeb2KO/Δex7 | Abnormal craniofacial, hearth and melanocyte development and defects in the PNS of the gastrointestinal tract and sympatho-adrenal lineage | van de Putte et al., 2007 [8] |
| Nestin-Cre+/−; Zeb2KO/Δex7 | Defects in cortical layering and in interneuron migration | Seuntjens et al., 2009 [47] |
| Nex-Cre+/−; Zeb2KO/Δex7 | Defects in cortical layering | Seuntjens et al., 2009 [47] |
| Nex-Cre+/−; Zeb2KO/Δex7; Ntf3−/− | Defects in cortical layering | Seuntjens et al., 2009 [47] |
| Olig1-Cre+/−; Zeb2Δex7/Δex7 | Defects in the maturation of precursor cells to oligodendrocytes and impaired myelin formation | Weng et al., 2012 [62] |
| Nkx2.1-Cre+/−; Zeb2KO/Δex7 | Defects in GABAergic interneuron migration | McKinsey et al., 2013, van den Berghe et al., 2013 [63,64] |
| Gsh2-Cre+/−; Zeb2KO/Δex7 | Defects in GABAergic interneuron migration and seizures | van den Berghe et al., 2013 [64] |
| Dlx5/6-Cre+/−; Zeb2KO/Δex7 | Defects in GABAergic interneuron migration | van den Berghe et al., 2013 [64] |
| Zeb2Δex7/+; pure C57BL/6N | Craniofacial abnormalities, defective corpus callosum formation, decreased numbers of parvalbumin interneurons in the cortex, reduced motor activity, increased anxiety, and impaired sociability | Takagi et al., 2015 [65] |
| Nex-Cre+/−; Zeb2+/Δex7 | Defects in axonal growth and ipsilateral intracortical collateral formation | Srivatsa et al., 2015 [66] |
| Dhh-Cre+/−; Zeb2Δex7/Δex7 | Arrest of Schwann Cell differentiation during peripheral nerve development and inhibition of remyelination after injury | Wu et al., 2016, Quintes et al., 2016 [67,68] |
| Dhh-Cre; Zeb2Δex7/Δex7; EdnrbΔ/Δ Dhh-Cre; Zeb2Δex7/Δex7; Hey2Δ/Δ | More mature axon-Schwann Cell units | Quintes et al., 2016 [67] |
| Nestin-Cre+/−; Zeb2KO/Δex7 | Increased BMP/Smad dependent axon growth and dopaminergic hyperinnervation in the striatum | Hegarty et al., 2017 [69] |
| Gsh2-Cre+/−; Zeb2KO/Δex7 | Defects in differentiation and maturation of olfactory bulb interneurons | Deryckere et al., 2020 [30] |
| Gfap-CreERT2; Zeb2Δex7/Δex7 | Larger lesions, and delays recovery of motor function after spinal cord injury or ischemic stroke | Vivinetto et al., 2020 [70] |
| Nex-Cre; Zeb2Δex7/Δex7 | Decreased expression of excitatory receptors and an impaired Ca2+ signaling | Turovskaya et al., 2020 [71] |
| Zeb2+/−; Sox10+/− | Defects in ENS | Stanchina et al., 2010 [59] |
| Zeb2KO/+; Ednrbs Zeb2KO/+; Edn3ls | Severe enteric anomalies and increased neuronal differentiation | Watanabe et al., 2017 [72] |
| Zeb2+/KO | Reduced pain response, defects in nociceptive transduction signals | Jeub et al., 2011 [73] |
| Zeb2+/KO | Reduced pain response, defects in DRG neuron development | Pradier et al., 2013 [74] |
| Brn4-Cre+/−; Zeb2KO/Δex7 | Defects in visceral motor neurons | Roy et al., 2012 [75] |
| Tyr-Cre; Zeb2Δex7/Δex7 | Defects in melanoblast migration and melanocyte differentiation | Denecker et al., 2014 [76] |
| Pax6(Lens)-Cre;ZeΔex7/Δex7 Pax6(LP)-Cre;Zeb2Δex7/Δex7 | Defects in vesicle lens closure and defects in lens fiber maturation | Yoshimoto et al., 2005 [77] |
| MLR10-Cre; Zeb2Δex7/Δex7 | Defects in coordinated cell migration, cataract formation and abnormalities in fiber cell organization in the lens | Manthey et al., 2014 [78] |
| α-Cre; Zeb2Δex7/Δex7 | Defects in cell numbers of various neuronal and glial cell types in the retina | Menuchin-Lasowski et al., 2016 [79] |
| Six3-Cre; Zeb2Δex7/Δex7 | Loss of non-photoreceptor cells, switch in cell fate to photoreceptor cells by retinal progenitors and increased apoptosis | Wei et al., 2019 [80] |
| Nrc1iCre; Zeb2Δex7/Δex7 | Impaired NK cell maturation, survival and bone marrow exit | Van Helden et al., 2015 |
| Nrc1iCre; R26-Zeb2Tg | Decreased NK cells in the bone marrow and an increase in mature NK cells in the spleen and bone marrow | Van Helden et al., 2015 [39] |
| αMHC-Cre; Zeb2Δex7/Δex7 | Impaired cardiac contractility and infarct healing post-myocardial infarction | Gladka et al., 2021 [81] |
| αMHC-Cre-R26Zeb2OE | Improved cardiomyocyte survival and cardiac function | Gladka et al., 2021 [81] |
| Cdh5-CreERT2; Zeb2Δex7/Δex7 | Expanded liver vasculature and irregularities in the angioarchitecture | De Haan et al., 2021 [82] |
| Cdh5-CreERT2; R26-Zeb2OE | Reduced vascularity and attenuated CCl4-induced liver fibrosis | De Haan et al., 2021 [82] |
| Tyr-CreERT2; Zeb2Δex7/Δexand Tyr-NRAS p53 | Decreased outgrowth of primary melanomas | Bruneel et al., 2020 [83] |
| R26-Zeb2OE/OEiresGFP | Increased proliferation and growth of primary and secondary melanomas | Bruneel et al., 2020 [83] |
| Model System | Location | Activity | References |
|---|---|---|---|
| Rat | rChr3: 26822763-26823523 | Post-natal kidney development | El-Kasti et al., 2012 [121] |
| Mouse | chr2:43,978,103-43,978,294 | GABA-ergic interneurons in developing subpallium | McKinsey et al., 2013 [63] |
| Zebrafish | Zeb2#e2: intron, Chr2, 14518542-14518630 | Notochord | Bar-Yaacov et al., 2019 [122] |
| Zebrafish | Zeb2#e3: intron, Chr2, 145188070-145189835 | Mid/hindbrain, spinal cord, forebrain | Bar-Yaacov et al., 2019 [122] |
| Zebrafish | Zeb2#e4: intron: Chr2, 145196296-145197640 | Notochord, non-specific neurons | Bar-Yaacov et al., 2019 [122] |
| Zebrafish | Zeb2#e5: intron, Chr2, 145201196-145202221 | Mid/hindbrain, somatic muscles, spinal cord | Bar-Yaacov et al., 2019 [122] |
| Zebrafish | Zeb2#e6: intron, Chr2, 145209727-145210776 | Trigeminal-like ganglia, somatic muscles | Bar-Yaacov et al., 2019 [122] |
| Zebrafish | Zeb2#e7: intron, Chr2, 145215740-145216978 | Trigeminal-like ganglia | Bar-Yaacov et al., 2019 [122] |
| Zebrafish | Zeb2#e12: intron, Chr2, 145265457-145266567 | Notochord | Bar-Yaacov et al., 2019 [122] |
| Zebrafish | Zeb2#e13: intron, Chr2, 145267933-145268902 | Somatic muscles | Bar-Yaacov et al., 2019 [122] |
| Zebrafish | Zeb2#e14: intron: Chr2, 145272461-145274126 | CNS | Bar-Yaacov et al., 2019 [122] |
| Human iPSCs | E1: Chr2:145764483–145765504 | NPC differentiation | Birkhoff et al., 2020 [123] |
| Human iPSCs | E2: Chr2:145769677–145770210 | NPC differentiation | Birkhoff et al., 2020 [123] |
| Human iPSCs | E3: Chr2:145779965– 145780193 | NPC differentiation | Birkhoff et al., 2020 [123] |
| miRNA | Regulation | References |
|---|---|---|
| miRNA-192 | TGFβ-induced collagen expression, and diabetic kidney glomeruli | Kato et al., 2007 [132] |
| miRNA-200 family | EMT | Bracken et al., 2008; Christoffersen et al., 2007; Gregory et al., 2008; Perdigão-Henriques et al., 2016 [125,127,129,130] |
| EMT and cancer cell migration | Korpal et al., 2008 [126] | |
| Epithelial phenotype of cancer cells | Park et al., 2008 [136] | |
| miRNA-200 | EMT in iPSCs | Wang et al., 2013 [128] |
| miRNA-205 | Renal carcinoma | Chen et al., 2014 [137] |
| miRNA-9 | Rat brain cortical development | Kropivsek et al., 2014 [131] |
| miRNA-145 | EMT and stem cell properties in prostate cancer | Ren et al., 2014 [135] |
| miRNA-200 family | Post-natal forebrain neurogenesis | Beclin et al., 2016 [124] |
| miRNA-215 | Metastasis of colorectal cancer | Chen et al., 2017 [138] |
| miRNA-30a | Triple negative breast cancer aggressiveness | Di Gennaro et al., 2018 [139] |
| miR200 family | CD8+ cell fates | Guan et al., 2018 [140] |
| miRNA-200b | Migration and invasion of oral squamous cell carcinoma | Ren et al., 2018 [141] |
| miRNA-203 | Colon cancer liver metastasis | Wang et al., 2018a [142] |
| miRNA-146 | Expression and replication of Hepatitis B virus | Wang et al., 2018b [143] |
| miRNA-206 | Proliferation of renal clear cell carcinoma | Chen et al., 2019a [144] |
| miRNA-30a | Human nasopharyngeal carcinoma | Chen et al., 2019b [145] |
| miRNA-505 | Metastasis and EMT in cervical cancer | Feng et al., 2019 [146] |
| miRNA-1179 | Metastasis of hepatocellular carcinoma | Gao et al., 2019 [147] |
| miRNA-101 | Proliferation and invasion of osteosarcoma | Lin et al., 2019 [148] |
| miRNA130b-3p | Migration and invasion of non-small lung carcinoma | Qu et al., 2019 [149] |
| miRNA-940 | EMT in glioma cells | Xu et al., 2019 [150] |
| miRNA-155 | Migration and invasion of colorectal cancer cells | Yang et al., 2019a [133] |
| miRNA-145 | Senescence of activated hepatic stellate cells | Yang et al., 2019b [134] |
| miRNA-498 | Growth and metastasis of liver cancer | Zhang et al., 2019a [151] |
| miRNA-200c-3p | Tumor progression of prostate carcinoma | Zhang et al., 2019b [152] |
| miR-124, miRNA-203 | EMT renal carcinoma | Chen et al., 2020 [153] |
| miRNA-215-5p | EMT in podocytes | Jin et al., 2020 [154] |
| miRNA-200a | Proliferation in drug-induced gingival overgrowth | Lin et al., 2020 [155] |
| miRNA-200c-3p | Proliferation and migration of renal artery endothelial cells | Liu et al., 2020 [156] |
| miRNA-200b | Senescence and inflammatory responses in pulmonary emphysema | Shen et al., 2020 [157] |
| miRNA-138 | Progression of colorectal cancer | Yan et al., 2020 [158] |
| miRNA-140 | Progression of esophageal cancer | Yang et al., 2021 [159] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birkhoff, J.C.; Huylebroeck, D.; Conidi, A. ZEB2, the Mowat-Wilson Syndrome Transcription Factor: Confirmations, Novel Functions, and Continuing Surprises. Genes 2021, 12, 1037. https://doi.org/10.3390/genes12071037
Birkhoff JC, Huylebroeck D, Conidi A. ZEB2, the Mowat-Wilson Syndrome Transcription Factor: Confirmations, Novel Functions, and Continuing Surprises. Genes. 2021; 12(7):1037. https://doi.org/10.3390/genes12071037
Chicago/Turabian StyleBirkhoff, Judith C., Danny Huylebroeck, and Andrea Conidi. 2021. "ZEB2, the Mowat-Wilson Syndrome Transcription Factor: Confirmations, Novel Functions, and Continuing Surprises" Genes 12, no. 7: 1037. https://doi.org/10.3390/genes12071037
APA StyleBirkhoff, J. C., Huylebroeck, D., & Conidi, A. (2021). ZEB2, the Mowat-Wilson Syndrome Transcription Factor: Confirmations, Novel Functions, and Continuing Surprises. Genes, 12(7), 1037. https://doi.org/10.3390/genes12071037