
New RAD51 Inhibitors to Target Homologous Recombination in Human Cells

 and
and
Abstract
1. Introduction
2. Materials and Methods
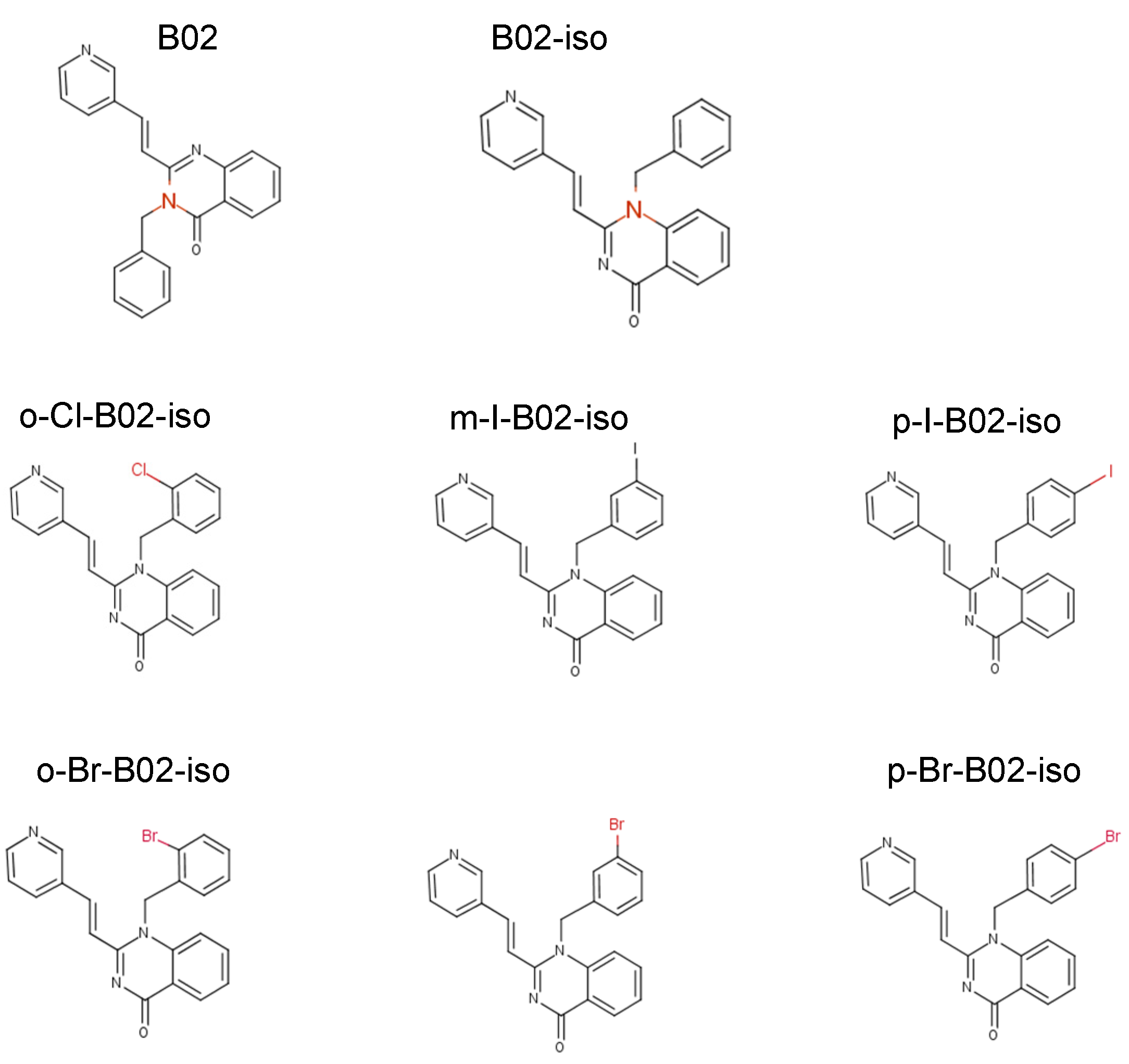
2.1. Chemicals
2.2. Cell Culture
2.3. Surface Plasmon Resonance (SPR) Assay
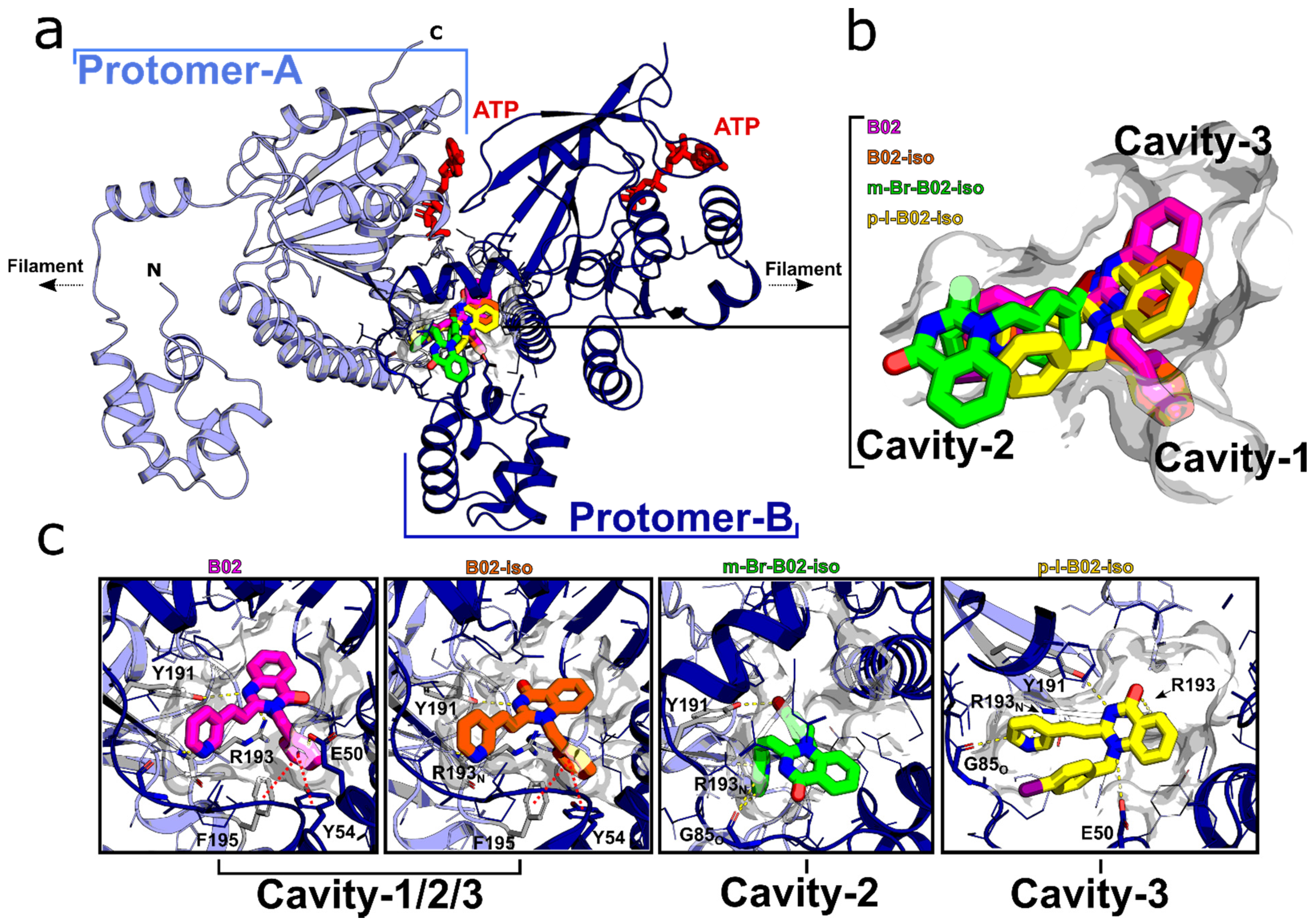
2.4. Docking Simulations
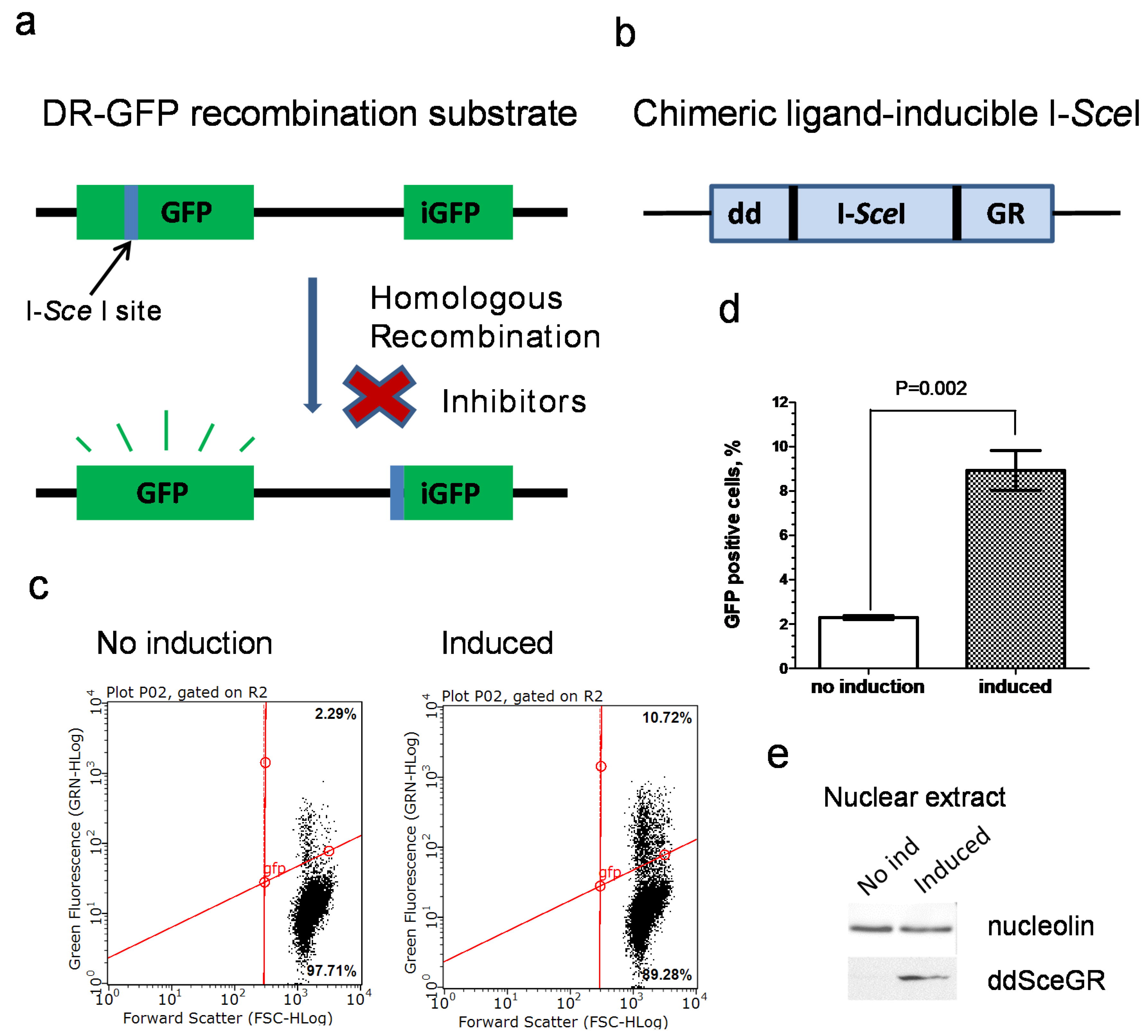
2.5. Construction of U-2 OS IndDR-GFP Cell Line
2.6. Inducible I-SceI Endonuclease DR-GFP Assay (IndDR-GFP)
2.7. Cell Viability Assay
2.8. RAD51 Foci Formation
2.9. Preparation of Nuclear and Whole-Cell Extracts
2.10. Western Blotting
2.11. EdU Cell Proliferation Assay
2.12. Cell Cycle Analysis
2.13. Statistical Analysis
3. Results
3.1. Development of an Inducible I-SceI DR-GFP Assay
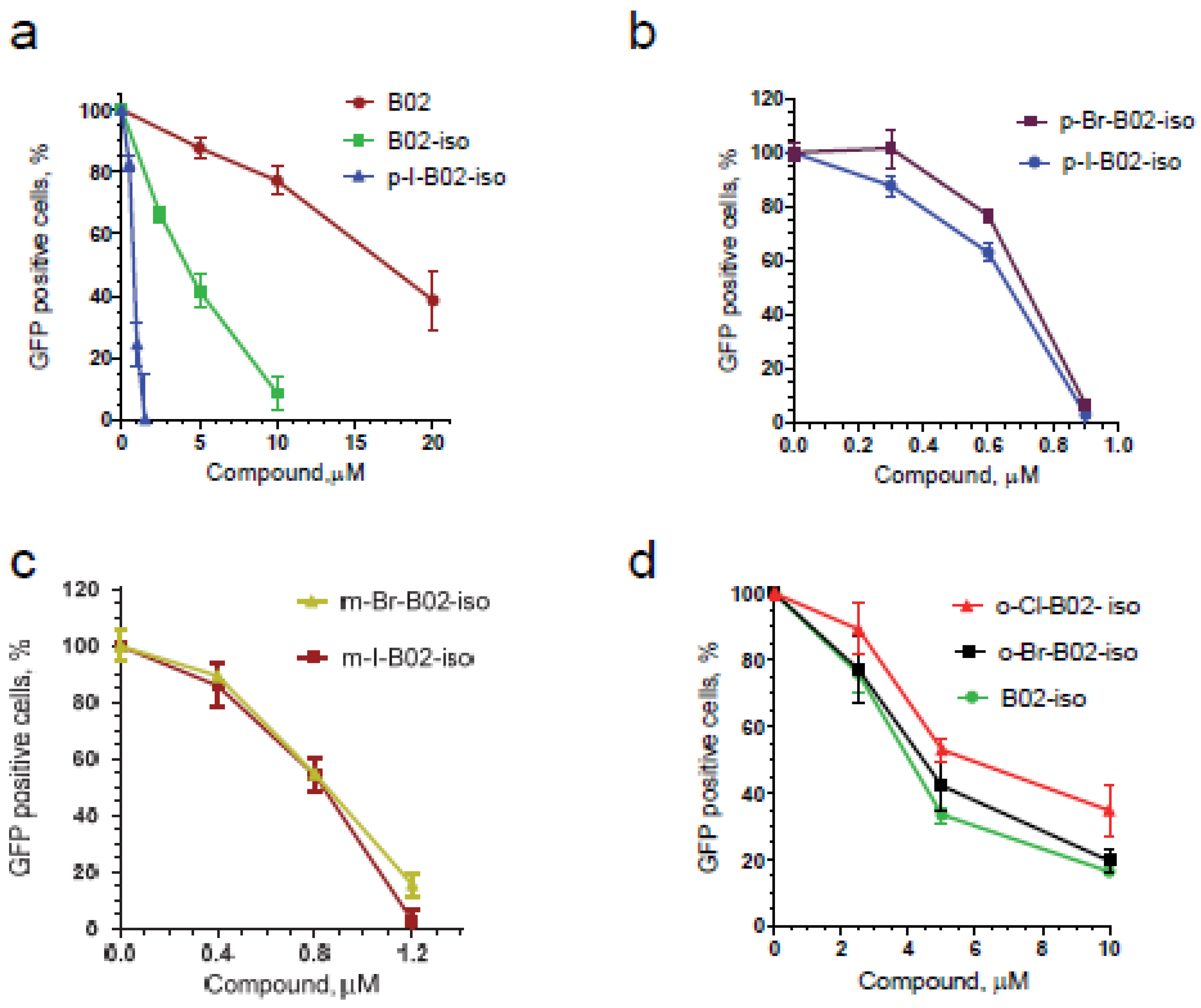
3.2. Testing Activity of New B02 Derivatives
3.3. B02-iso Analogs Bind Directly to RAD51 In Vitro
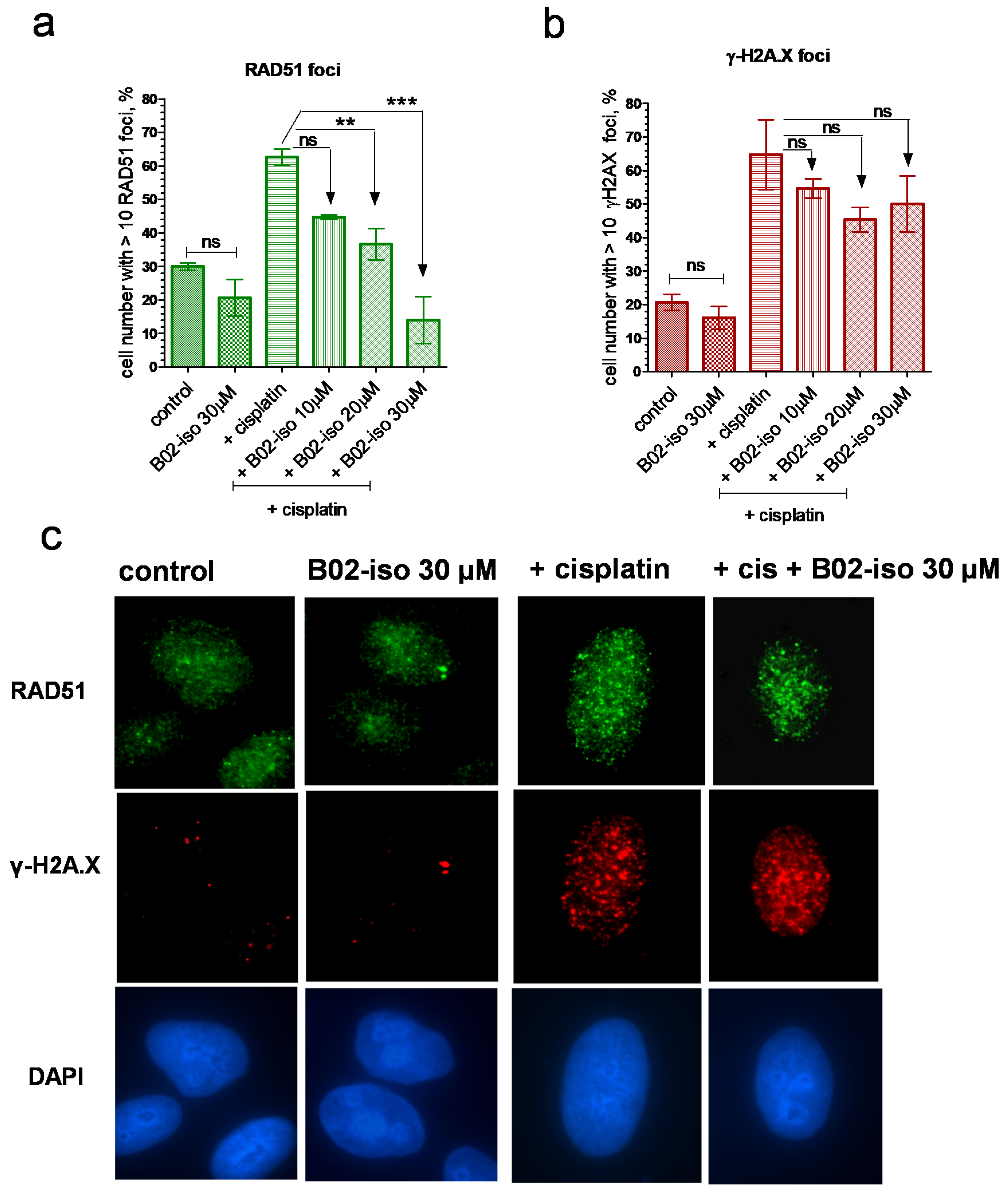
3.4. B02-iso Inhibits RAD51 Foci Formation in Human Cells
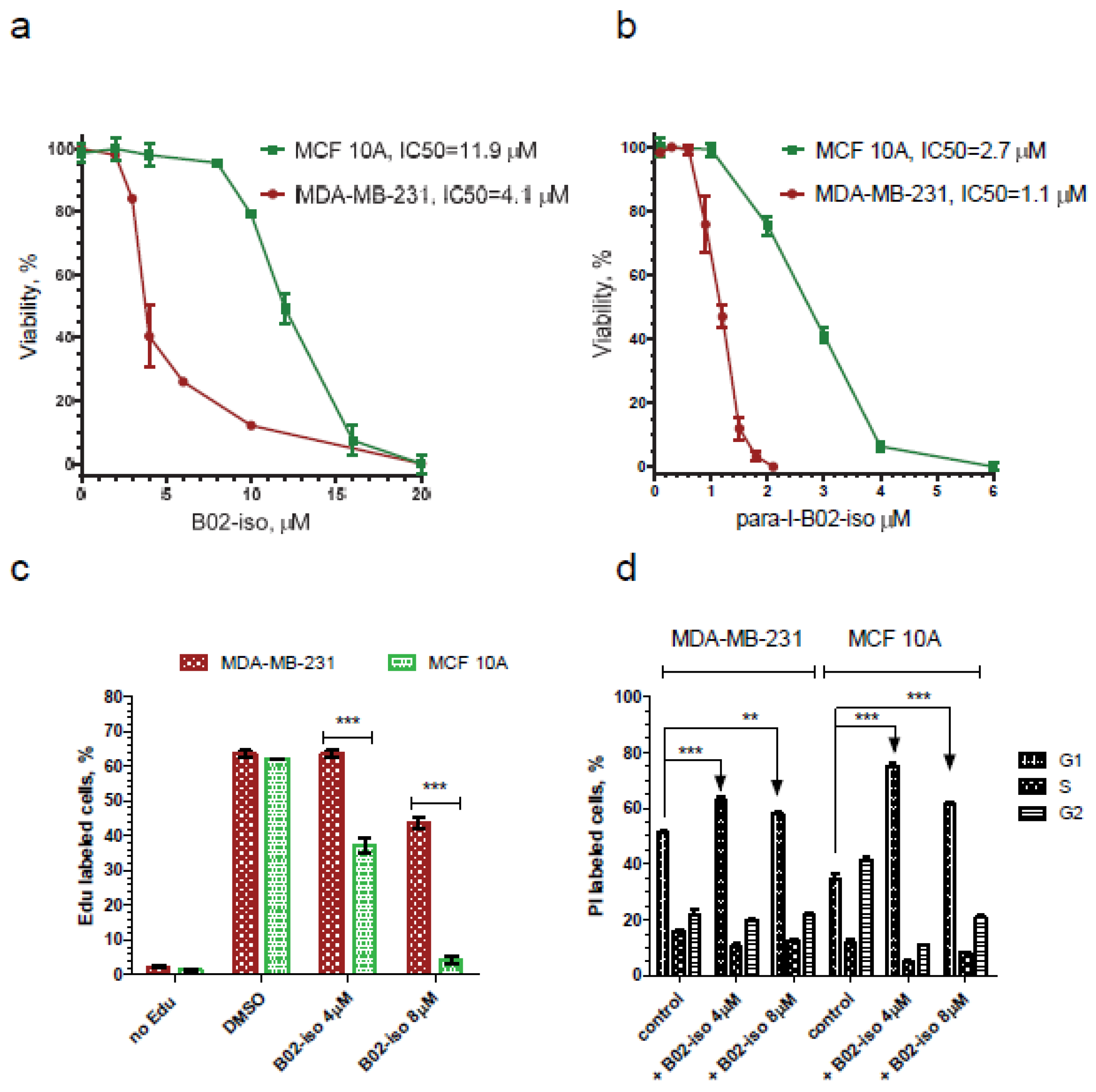
3.5. B02-iso Analogs Show Preferential Antiproliferative Effect in Cancer Cells
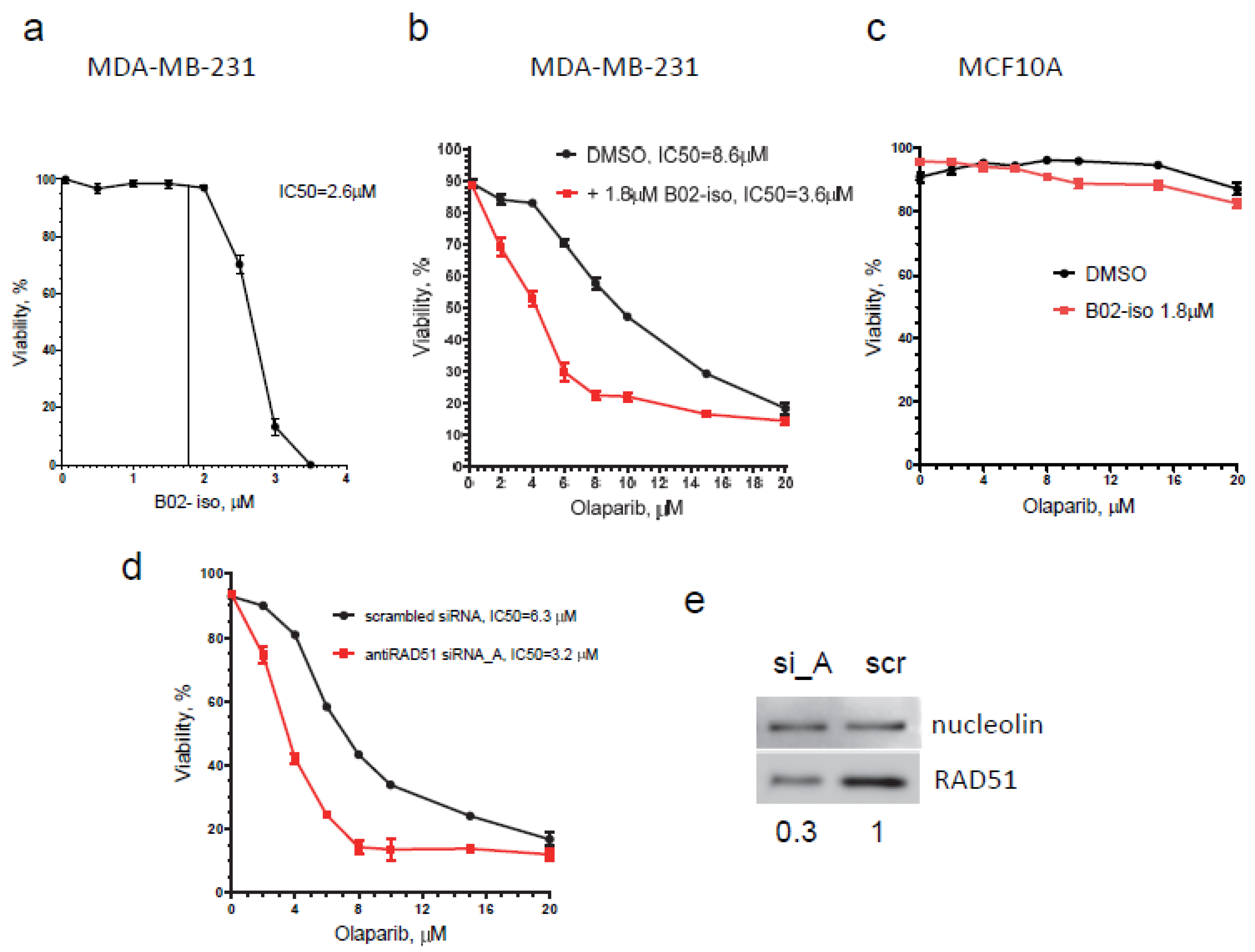
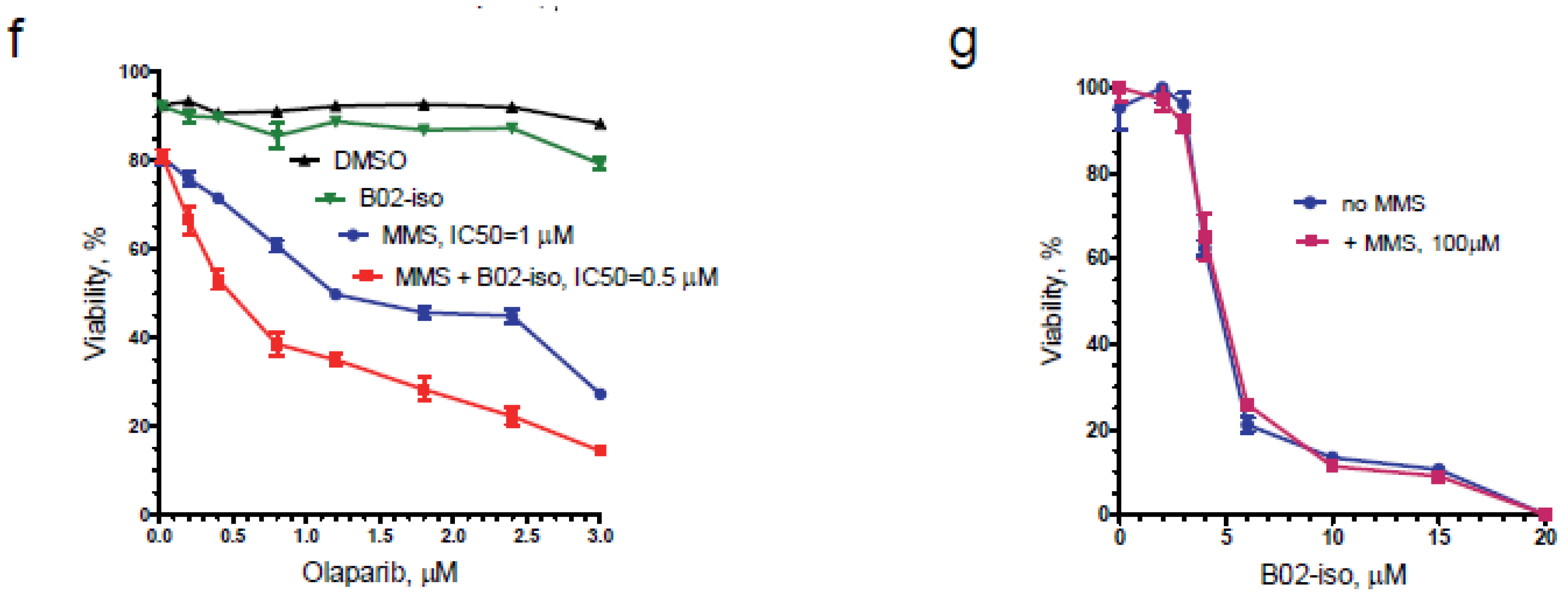
3.6. B02-iso Potentiates the Killing of Triple-Negative Breast Cancer Cells with Olaparib
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Ann. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef]
- Bell, J.C.; Kowalczykowski, S.C. Mechanics and Single-Molecule Interrogation of DNA Recombination. Ann. Rev. Biochem. 2016, 85, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 1994, 265, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Yu, X.; Shinohara, A.; Egelman, E.H. Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science 1993, 259, 1896–1899. [Google Scholar] [CrossRef]
- Benson, F.E.; Stasiak, A.; West, S.C. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J. 1994, 13, 5764–5771. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef]
- Davies, A.A.; Masson, J.Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Shivji, M.K.; Mukund, S.R.; Rajendra, E.; Chen, S.; Short, J.M.; Savill, J.; Klenerman, D.; Venkitaraman, A.R. The BRC repeats of human BRCA2 differentially regulate RAD51 binding on single- versus double-stranded DNA to stimulate strand exchange. Proc. Natl. Acad. Sci. USA 2009, 106, 13254–13259. [Google Scholar] [CrossRef] [PubMed]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Murayama, Y.; Kurokawa, Y.; Kanamaru, S.; Kokabu, Y.; Maki, T.; Mikawa, T.; Argunhan, B.; Tsubouchi, H.; Ikeguchi, M.; et al. Real-time tracking reveals catalytic roles for the two DNA binding sites of Rad51. Nat. Commun. 2020, 11, 2950. [Google Scholar] [CrossRef]
- Holloman, W.K.; Radding, C.M. Recombination promoted by superhelical DNA and the recA gene of Escherichia coli. Proc. Natl. Acad. Sci. USA 1976, 73, 3910–3914. [Google Scholar] [CrossRef]
- Haber, J.E. DNA Repair: The Search for Homology. Bioessays 2018, 40, e1700229. [Google Scholar] [CrossRef]
- Johnson, R.D.; Jasin, M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000, 19, 3398–3407. [Google Scholar] [CrossRef]
- Raderschall, E.; Stout, K.; Freier, S.; Suckow, V.; Schweiger, S.; Haaf, T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002, 62, 219–225. [Google Scholar]
- Maacke, H.; Jost, K.; Opitz, S.; Miska, S.; Yuan, Y.; Hasselbach, L.; Luttges, J.; Kalthoff, H.; Sturzbecher, H.W. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene 2000, 19, 2791–2795. [Google Scholar] [CrossRef]
- Maacke, H.; Opitz, S.; Jost, K.; Hamdorf, W.; Henning, W.; Kruger, S.; Feller, A.C.; Lopens, A.; Diedrich, K.; Schwinger, E.; et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int. J. Cancer 2000, 88, 907–913. [Google Scholar] [CrossRef]
- Barbano, R.; Copetti, M.; Perrone, G.; Pazienza, V.; Muscarella, L.A.; Balsamo, T.; Storlazzi, C.T.; Ripoli, M.; Rinaldi, M.; Valori, V.M.; et al. High RAD51 mRNA expression characterize estrogen receptor-positive/progesteron receptor-negative breast cancer and is associated with patient’s outcome. Int. J. Cancer 2011, 129, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.S.; Kuo, Y.H.; Chiu, Y.F.; Su, Y.C.; Lin, Y.W. Down-regulation of Rad51 expression overcomes drug resistance to gemcitabine in human non-small-cell lung cancer cells. J. Pharmacol. Exp. Ther. 2010, 335, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Du, L.Q.; Wang, Y.; Wang, H.; Cao, J.; Liu, Q.; Fan, F.Y. Knockdown of Rad51 expression induces radiation- and chemo-sensitivity in osteosarcoma cells. Med. Oncol. 2011, 28, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Wiegmans, A.P.; Al-Ejeh, F.; Chee, N.; Yap, P.Y.; Gorski, J.J.; Da Silva, L.; Bolderson, E.; Chenevix-Trench, G.; Anderson, R.; Simpson, P.T.; et al. Rad51 supports triple negative breast cancer metastasis. Oncotarget 2014, 5, 3261–3272. [Google Scholar] [CrossRef]
- Huang, F.; Mazin, A.V. Targeting the homologous recombination pathway by small molecule modulators. Bioorg. Med. Chem. Lett. 2014, 24, 3006–3013. [Google Scholar] [CrossRef][Green Version]
- Chen, Q.; Cai, D.; Li, M.; Wu, X. The homologous recombination protein RAD51 is a promising therapeutic target for cervical carcinoma. Oncol. Rep. 2017, 38, 767–774. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Budke, B.; Lv, W.; Kozikowski, A.P.; Connell, P.P. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? ChemMedChem 2016, 11, 2468–2473. [Google Scholar] [CrossRef]
- Budke, B.; Logan, H.L.; Kalin, J.H.; Zelivianskaia, A.S.; Cameron McGuire, W.; Miller, L.L.; Stark, J.M.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. RI-1: A chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012, 40, 7347–7357. [Google Scholar] [CrossRef]
- Budke, B.; Tueckmantel, W.; Miles, K.; Kozikowski, A.P.; Connell, P.P. Optimization of Drug Candidates That Inhibit the D-Loop Activity of RAD51. ChemMedChem 2019, 14, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, H.; Guo, X.E.; Qiu, X.L.; Hu, C.M.; Chamberlin, A.R.; Lee, W.H. Synthesis, molecular modeling, and biological evaluation of novel RAD51 inhibitors. Eur. J. Med. Chem. 2015, 96, 196–208. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, L.; Wu, G.; Konig, H.; Lin, X.; Li, G.; Qiu, X.L.; Chen, C.F.; Hu, C.M.; Goldblatt, E.; et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol. Med. 2013, 5, 353–365. [Google Scholar] [CrossRef]
- Bagnolini, G.; Milano, D.; Manerba, M.; Schipani, F.; Ortega, J.A.; Gioia, D.; Falchi, F.; Balboni, A.; Farabegoli, F.; De Franco, F.; et al. Synthetic Lethality in Pancreatic Cancer: Discovery of a New RAD51-BRCA2 Small Molecule Disruptor That Inhibits Homologous Recombination and Synergizes with Olaparib. J. Med. Chem. 2020, 63, 2588–2619. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Mazina, O.M.; Zentner, I.J.; Cocklin, S.; Mazin, A.V. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J. Med. Chem. 2012, 55, 3011–3020. [Google Scholar] [CrossRef]
- Huang, F.; Mazin, A.V. A Small Molecule Inhibitor of Human RAD51 Potentiates Breast Cancer Cell Killing by Therapeutic Agents in Mouse Xenografts. PLoS ONE 2014, 9, e100993. [Google Scholar] [CrossRef] [PubMed]
- Pletz, J.; Berg, B.; Rolf, B. A General and Direct Reductive Amination of Aldehydes and Ketones with Electron-Deficient Anilines. Synthesis 2016, 48, 8. [Google Scholar]
- Kabalka, G.W.; Deshpande, S.M.; Wadgaonkar, P.P.; Chatla, N. The Transformation of Nitriles into Amides Using Sodium Percarbonate. Synth. Commun. 2006, 20, 1445–1451. [Google Scholar] [CrossRef]
- Langhals, H.; Becherer, T.; Lindner, J.; Obermeier, A. The fluorescence labelling of primary amines with perylenetetracarboxdiimides. Eur. J. Org. Chem. 2007, 2007, 4328–4336. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarkian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 15. [Google Scholar] [CrossRef]
- Solis, F.J.; Wets, R.J.B. Minimization by Random Search Techniques. Math. Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]
- Bindra, R.S.; Goglia, A.G.; Jasin, M.; Powell, S.N. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013, 41, e115. [Google Scholar] [CrossRef][Green Version]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Dong, L.; Harris, J.M.; Khanna, K.K.; Al-Ejeh, F.; Fairlie, D.P.; Wiegmans, A.P.; Liu, L. Quinazolinone derivatives as inhibitors of homologous recombinase RAD51. Bioorg. Med. Chem. Lett. 2017, 27, 3096–3100. [Google Scholar] [CrossRef]
- Vydyam, P.; Dutta, D.; Sutram, N.; Bhattacharyya, S.; Bhattacharyya, M.K. A small-molecule inhibitor of the DNA recombinase Rad51 from Plasmodium falciparum synergizes with the antimalarial drugs artemisinin and chloroquine. J. Biol. Chem. 2019, 294, 8171–8183. [Google Scholar] [CrossRef] [PubMed]
- Haaf, T.; Golub, E.I.; Reddy, G.; Radding, C.M.; Ward, D.C. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA 1995, 92, 2298–2302. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, N.; Yao, W.; Li, S.; Ren, Z. RAD51 is a potential marker for prognosis and regulates cell proliferation in pancreatic cancer. Cancer Cell Int. 2019, 19, 356. [Google Scholar] [CrossRef]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Falchi, F.; Giacomini, E.; Masini, T.; Boutard, N.; Di Ianni, L.; Manerba, M.; Farabegoli, F.; Rossini, L.; Robertson, J.; Minucci, S.; et al. Synthetic Lethality Triggered by Combining Olaparib with BRCA2-Rad51 Disruptors. ACS Chem. Biol. 2017, 12, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Heacock, M.L.; Stefanick, D.F.; Horton, J.K.; Wilson, S.H. Alkylation DNA damage in combination with PARP inhibition results in formation of S-phase-dependent double-strand breaks. DNA Repair 2010, 9, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Endo, S.; Chen, Z.; Qi, H.; Watanabe, G.; Chiba, N. Evaluation of site-specific homologous recombination activity of BRCA1 by direct quantitation of gene editing efficiency. Sci. Rep. 2019, 9, 1644. [Google Scholar] [CrossRef]
- Chatterjee, G.; Jimenez-Sainz, J.; Presti, T.; Nguyen, T.; Jensen, R.B. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res. 2016, 44, 5256–5270. [Google Scholar] [CrossRef]
- Goglia, A.G.; Delsite, R.; Luz, A.N.; Shahbazian, D.; Salem, A.F.; Sundaram, R.K.; Chiaravalli, J.; Hendrikx, P.J.; Wilshire, J.A.; Jasin, M.; et al. Identification of Novel Radiosensitizers in a High-Throughput, Cell-Based Screen for DSB Repair Inhibitors. Mol. Cancer Ther. 2015, 14, 326–342. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Zhao, J.; Kumar, S.; Chakraborty, C.; Talluri, S.; Munshi, N.C.; Shammas, M.A. RAD51 Inhibitor Reverses Etoposide-Induced Genomic Toxicity and Instability in Esophageal Adenocarcinoma Cells. Arch. Clin. Toxicol. 2020, 2, 3–9. [Google Scholar]
- Shammas, M.A.; Shmookler Reis, R.J.; Koley, H.; Batchu, R.B.; Li, C.; Munshi, N.C. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 2009, 113, 2290–2297. [Google Scholar] [CrossRef]
- Pal, J.; Bertheau, R.; Buon, L.; Qazi, A.; Batchu, R.B.; Bandyopadhyay, S.; Ali-Fehmi, R.; Beer, D.G.; Weaver, D.W.; Shmookler Reis, R.J.; et al. Genomic evolution in Barrett’s adenocarcinoma cells: Critical roles of elevated hsRAD51, homologous recombination and Alu sequences in the genome. Oncogene 2011, 30, 3585–3598. [Google Scholar] [CrossRef]
- Bishop, A.J.; Schiestl, R.H. Role of homologous recombination in carcinogenesis. Exp. Mol. Pathol. 2003, 74, 94–105. [Google Scholar] [CrossRef]
- Klein, H.L. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 2008, 7, 686–693. [Google Scholar] [CrossRef]
- Hine, C.M.; Li, H.; Xie, L.; Mao, Z.; Seluanov, A.; Gorbunova, V. Regulation of Rad51 promoter. Cell Cycle 2014, 13, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Yata, K.; Lloyd, J.; Maslen, S.; Bleuyard, J.Y.; Skehel, M.; Smerdon, S.J.; Esashi, F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 2012, 45, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef]
- Saeki, H.; Jogo, T.; Kawazoe, T.; Kamori, T.; Nakaji, Y.; Zaitsu, Y.; Fujiwara, M.; Baba, Y.; Nakamura, T.; Iwata, N.; et al. RAD51 Expression as a Biomarker to Predict Efficacy of Preoperative Therapy and Survival for Esophageal Squamous Cell Carcinoma: A Large-cohort Observational Study (KSCC1307). Ann. Surg. 2020. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Zhang, Z.T. Genetic Polymorphisms in the RAD51 Gene with a Risk of Head and Neck Cancer and Esophageal Cancer: A Meta-Analysis. Int. J. Genom. 2019, 2019, 2789035. [Google Scholar] [CrossRef]
- Grundy, M.K.; Buckanovich, R.J.; Bernstein, K.A. Regulation and pharmacological targeting of RAD51 in cancer. NAR Cancer 2020, 2, zcaa024. [Google Scholar] [CrossRef]
- Roberti, M.; Schipani, F.; Bagnolini, G.; Milano, D.; Giacomini, E.; Falchi, F.; Balboni, A.; Manerba, M.; Farabegoli, F.; De Franco, F.; et al. Rad51/BRCA2 disruptors inhibit homologous recombination and synergize with olaparib in pancreatic cancer cells. Eur. J. Med. Chem. 2019, 165, 80–92. [Google Scholar] [CrossRef]
- Raderschall, E.; Golub, E.I.; Haaf, T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 1921–1926. [Google Scholar] [CrossRef]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutierrez-Enriquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Malcikova, J.; Tichy, B.; Damborsky, J.; Kabathova, J.; Trbusek, M.; Mayer, J.; Pospisilova, S. Analysis of the DNA-binding activity of p53 mutants using functional protein microarrays and its relationship to transcriptional activation. Biol. Chem. 2010, 391, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.S.; Trovao, F.; Andrade Pinheiro, B.; Freire, F.; Gomes, S.; Oliveira, C.; Domingues, L.; Romao, M.J.; Saraiva, L.; Carvalho, A.L. The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding. Int. J. Mol. Sci. 2018, 19, 1184. [Google Scholar] [CrossRef]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Kuerbitz, S.J. Control of G1 arrest after DNA damage. Environ. Health Perspect. 1993, 101 (Suppl. 5), 55–58. [Google Scholar] [CrossRef]
- Moxley, A.H.; Reisman, D. Context is key: Understanding the regulation, functional control, and activities of the p53 tumour suppressor. Cell Biochem. Funct. 2020, 39, 235–247. [Google Scholar] [CrossRef]
- Aubry, A.; Pearson, J.D.; Huang, K.; Livne-Bar, I.; Ahmad, M.; Jagadeesan, M.; Khetan, V.; Ketela, T.; Brown, K.R.; Yu, T.; et al. Functional genomics identifies new synergistic therapies for retinoblastoma. Oncogene 2020, 39, 5338–5357. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: The First Synthetic Lethality Targeted therapy. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 2018, 24, 3513–3527.e7. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 8. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Bugreev, D.V.; Rossi, M.J.; Mazin, A.V. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 2011, 39, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 | KD |
|---|---|---|
| B02 | 17.70 ± 3.89 µM | 14.6 ± 7.8 µM |
| B02-iso | 4.30 ± 0.75 µM | 14.6 ± 6.2 µM |
| p-I-B02-iso | 0.72 ± 0.07 µM | 1.4 ± 0.6 µM |
| p-Br-B02-iso | 0.80 ± 0.10 µM | ND |
| m-Br-B02-iso | 0.90 ± 0.18 µM | 8.4 ± 1.8 µM |
| m-I-B02-iso | 0.86 ± 0.02 µM | ND |
| o-Cl-B02-iso | 5.56 ± 0.49 µM | ND |
| o-Br-B02-iso | 4.60 ± 1.28 µM | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shkundina, I.S.; Gall, A.A.; Dick, A.; Cocklin, S.; Mazin, A.V. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes 2021, 12, 920. https://doi.org/10.3390/genes12060920
Shkundina IS, Gall AA, Dick A, Cocklin S, Mazin AV. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes. 2021; 12(6):920. https://doi.org/10.3390/genes12060920
Chicago/Turabian StyleShkundina, Irina S., Alexander A. Gall, Alexej Dick, Simon Cocklin, and Alexander V. Mazin. 2021. "New RAD51 Inhibitors to Target Homologous Recombination in Human Cells" Genes 12, no. 6: 920. https://doi.org/10.3390/genes12060920
APA StyleShkundina, I. S., Gall, A. A., Dick, A., Cocklin, S., & Mazin, A. V. (2021). New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes, 12(6), 920. https://doi.org/10.3390/genes12060920