
Cytogenetic and Array-CGH Characterization of a Simple Case of Reciprocal t(3;10) Translocation Reveals a Hidden Deletion at 5q12


 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient
2.2. Cytogenetic Analyses
2.3. Array-CGH Analysis
2.4. Fluorescence In Situ Hybridization (FISH) Analysis
2.5. Repetitive Elements Analysis
3. Results
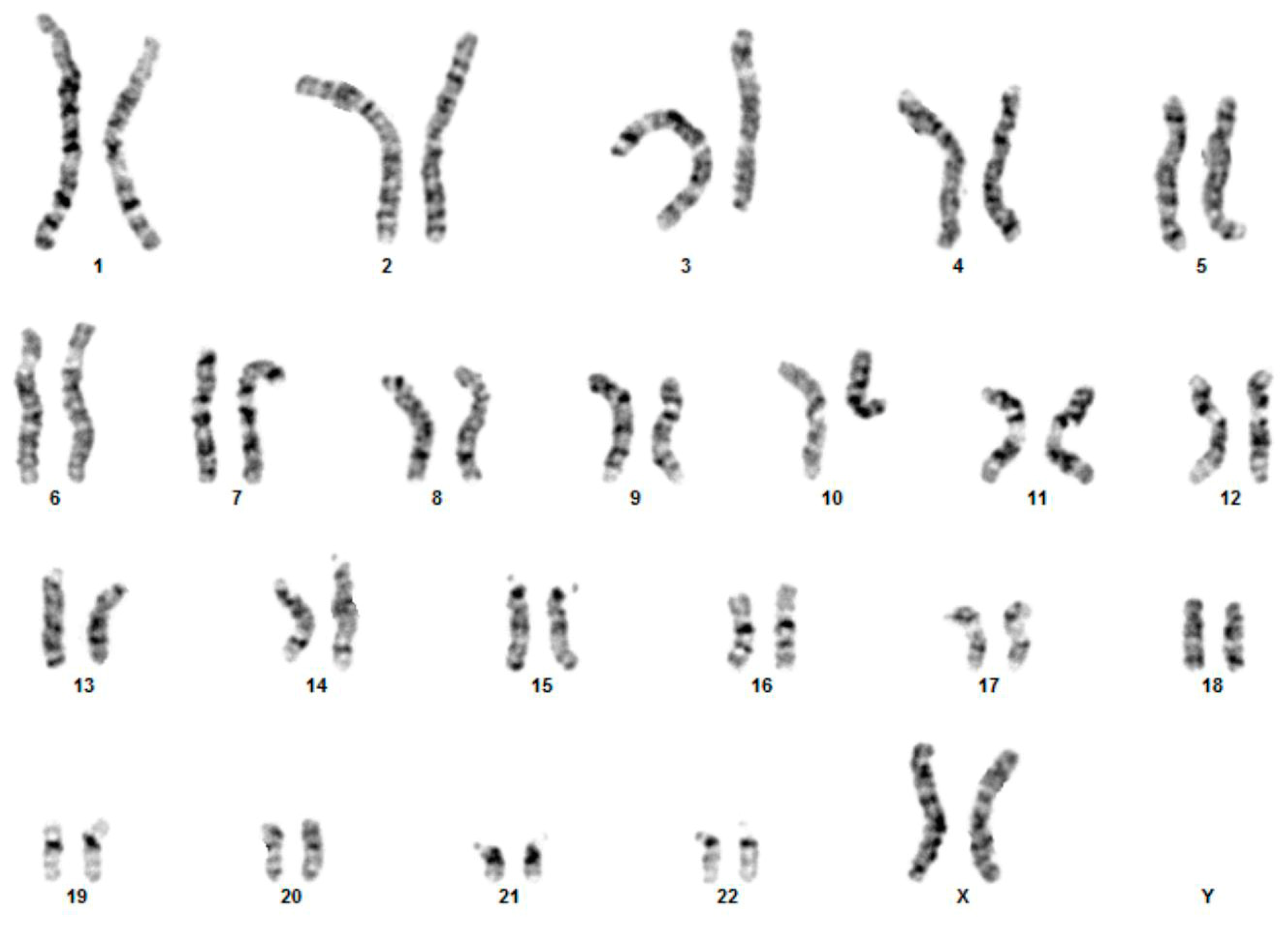
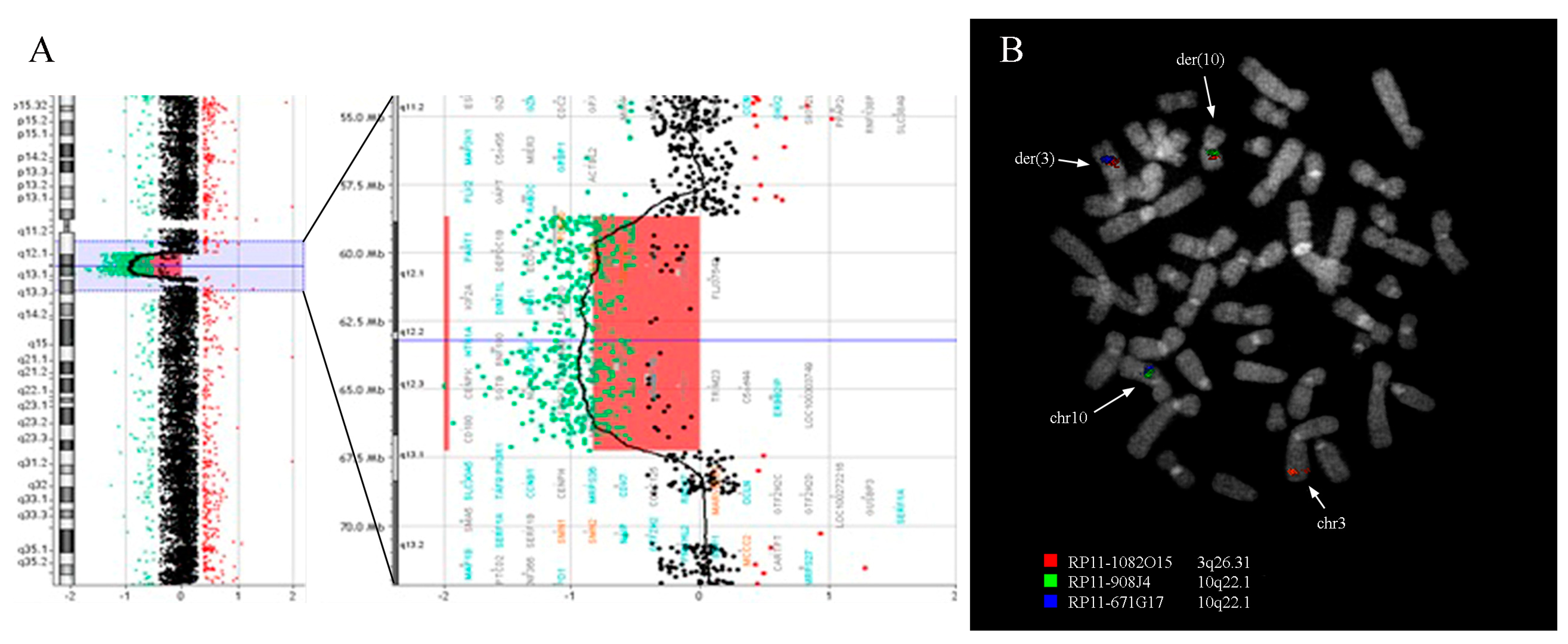
3.1. Cytogenetic and Array-CGH Analysis
3.2. Fluorescence In Situ Hybridization Analysis
3.3. Bioinformatic Analysis of Breakpoint Regions
3.4. Gene Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lupski, J.R. Genomic disorders ten years on. Genome Med. 2009, 1, 1–11. [Google Scholar] [CrossRef]
- Jaillard, S.; Andrieux, J.; Plessis, G.; Krepischi, A.C.; Lucas, J.; David, V.; Le Brun, M.; Bertola, D.R.; David, A.; Belaud-Rotureau, M.-A.; et al. 5q12.1 deletion: Delineation of a phenotype including mental retardation and ocular defects. Am. J. Med. Genet. Part A 2011, 155, 725–731. [Google Scholar] [CrossRef]
- Cetin, Z.; Yakut, S.; Clark, O.A.; Mihci, E.; Berker, S.; Luleci, G. A 5q12.1–5q12.3 microdeletion in a case with a balanced exceptional complex chromosomal rearrangement. Gene 2013, 516, 176–180. [Google Scholar] [CrossRef]
- Lindstrand, A.; Grigelioniene, G.; Nilsson, D.; Pettersson, M.; Hofmeister, W.; Anderlid, B.-M.; Kant, S.G.; Ruivenkamp, C.A.L.; Gustavsson, P.; Valta, H.; et al. Different mutations inPDE4Dassociated with developmental disorders with mirror phenotypes. J. Med. Genet. 2013, 51, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Holder, J.L.; Cheung, S.-W. Refinement of the postnatal growth restriction locus of chromosome 5q12-13 deletion syndrome. Am. J. Med. Genet. Part A 2015, 167, 2737–2741. [Google Scholar] [CrossRef] [PubMed]
- Gnan, C.; Franzoni, A.; Baldan, F.; Passon, N.; Damante, G.; Russo, P.D. Familial 5q12.3 Microdeletion: Evidence for a Locus Associated with Epilepsy. Mol. Syndr. 2017, 8, 98–102. [Google Scholar] [CrossRef]
- Specchia, G.; Albano, F.; Anelli, L.; Storlazzi, C.T.; Zagaria, A.; Liso, A.; Pannunzio, A.; Pastore, D.; Mestice, A.; Greco, G.; et al. Derivative Chromosome 9 Deletions in Chronic Myeloid Leukemia are Associated with Loss of Tumor Suppressor Genes. Leuk. Lymphoma 2004, 45, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; Minervini, A.; Rossi, A.R.; Specchia, G. Decreased TET2 gene expression during chronic myeloid leukemia progression. Leuk. Res. 2011, 35, e220–e222. [Google Scholar] [CrossRef]
- Ichtchenko, K.; Hata, Y.; Nguyen, T.; Ullrich, B.; Missler, M.; Moomaw, C.; Südhof, T.C. Neuroligin 1: A splice site-specific ligand for β-neurexins. Cell 1995, 81, 435–443. [Google Scholar] [CrossRef]
- Katzman, A.; Alberini, C.M. NLGN1 and NLGN2 in the prefrontal cortex: Their role in memory consolidation and strengthening. Curr. Opin. Neurobiol. 2018, 48, 122–130. [Google Scholar] [CrossRef]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef]
- Nakanishi, M.; Nomura, J.; Ji, X.; Tamada, K.; Arai, T.; Takahashi, E.; Bućan, M.; Takumi, T. Functional significance of rare neuroligin 1 variants found in autism. PLoS Genet. 2017, 13, e1006940. [Google Scholar] [CrossRef]
- Chahine, L.; Abou-Khalil, B.; Siren, A.; Andermann, F.; Hedera, P.; Ge, Q.; Andermann, E.; Pandolfo, M. A new locus for familial temporal lobe epilepsy on chromosome 3q. Epilepsy Res. 2013, 106, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Giri, M.; Xia, Z.; Subedi, Y.N.; Li, Y. Genetic and epigenetic mechanisms of epilepsy: A review. Neuropsychiatr. Dis. Treat. 2017, 13, 1841–1859. [Google Scholar] [CrossRef]
- Li, W.; Pozzo-Miller, L. Dysfunction of the corticostriatal pathway in autism spectrum disorders. J. Neurosci. Res. 2020, 98, 2130–2147. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-M.; Stenson, P.D.; Cooper, D.N.; Férec, C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Qual. Life Res. 2005, 117, 411–427. [Google Scholar] [CrossRef]
- Gasior, S.; Wakeman, T.P.; Xu, B.; Deininger, P.L. The Human LINE-1 Retrotransposon Creates DNA Double-strand Breaks. J. Mol. Biol. 2006, 357, 1383–1393. [Google Scholar] [CrossRef]
- Mahadevaiah, S.K.; Turner, J.M.; Baudat, F.; Rogakou, E.P.; De Boer, P.; Blanco-Rodríguez, J.; Jasin, M.; Keeney, S.; Bonner, W.M.; Burgoyne, P.S. Recombinational DNA double-strand breaks in mice precede synapsis. Nat. Genet. 2001, 27, 271–276. [Google Scholar] [CrossRef]
- Michot, C.; LE Goff, C.; Goldenberg, A.; Abhyankar, A.; Klein, C.; Kinning, E.; Guerrot, A.-M.; Flahaut, P.; Duncombe, A.; Baujat, G.; et al. Exome Sequencing Identifies PDE4D Mutations as Another Cause of Acrodysostosis. Am. J. Hum. Genet. 2012, 90, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.-L.C.; Richard, F.J.; Kuo, W.-P.; D’Ercole, A.J.; Conti, M. Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc. Natl. Acad. Sci. USA 1999, 96, 11998–12003. [Google Scholar] [CrossRef] [PubMed]
- Thauvin-Robinet, C.; Auclair, M.; Duplomb, L.; Caron-Debarle, M.; Avila, M.; St-Onge, J.; Le Merrer, M.; Le Luyer, B.; Héron, D.; Mathieu-Dramard, M.; et al. PIK3R1 Mutations Cause Syndromic Insulin Resistance with Lipoatrophy. Am. J. Hum. Genet. 2013, 93, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ibn-Salem, J.; Köhler, S.; Love, M.I.; Chung, H.-R.; Huang, N.; Hurles, M.E.; Haendel, M.; Washington, N.L.; Smedley, D.; Mungall, C.J.; et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 2014, 15, 1–16. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| BAC Clones | GRCh38/hg38 Coordinates | Cytogenetic Band | FISH Mapping |
|---|---|---|---|
| RP11-641D5 | chr3:169447867-169477052 | 3q26.2 | chr3; der(3) |
| RP11-1142D2 | chr3:171349615-171504931 | 3q26.31 | chr3; der(3) |
| RP11-946M22 | chr3:173538354-173727586 | 3q26.31 | chr3; der(3) |
| RP11-1082O15 | chr3:173727590-173911910 | 3q26.31 | chr3; der(3); der(10) |
| RP11-1082C2 | chr3:173727583-173911912 | 3q26.31 | chr3; der(10) |
| RP11-717I16 | chr3:173865121-174018111 | 3q26.31 | chr3; der(10) |
| RP11-910H14 | chr3:174788143-174993161 | 3q26.31 | chr3; der(10) |
| RP11-1072A11 | chr3:175242524-175426450 | 3q26.31 | chr3; der(10) |
| RP11-1026C21 | chr3:175742862-175922047 | 3q26.31 | chr3; der(10) |
| RP11-671L21 | chr3:178934872-179103061 | 3q26.32 | chr3; der(10) |
| RP11-1030A12 | chr3:182598218-182796672 | 3q26.33-q27.1 | chr3; der(10) |
| RP11-1149L21 | chr10:49879596-50032584 | 10q11.23 | chr10; der(10) |
| RP11-1053M8 | chr10:63425441-63600635 | 10q21.3 | chr10; der(10) |
| RP11-757B19 | chr10:64486402-64649519 | 10q21.3 | chr10; der(10) |
| RP11-1044J13 | chr10:66778400-66962952 | 10q21.3 | chr10; der(10) |
| RP11-705E19 | chr10:68313547-68476410 | 10q21.3 | chr10; der(10) |
| RP11-718E13 | chr10:68512130-68694593 | 10q21.3 | chr10; der(10) |
| RP11-1133N9 | chr10:68893282-69042226 | 10q22.1 | chr10; der(10) |
| RP11-960P24 | chr10:69042237-69218463 | 10q22.1 | chr10; der(10) |
| RP11-625N12 | chr10:69153908-69438414 | 10q22.1 | chr10; der(10) |
| RP11-876K24 | chr10:69433272-69603807 | 10q22.1 | chr10; der(10) |
| RP11-846C5 | chr10:69607071-69786886 | 10q22.1 | chr10; der(10) |
| RP11-1149M15 | chr10:69806806-69952093 | 10q22.1 | chr10; der(10) |
| RP11-826A6 | chr10:69931863-70103844 | 10q22.1 | chr10; der(10) |
| RP11-691F7 | chr10:70153017-70350532 | 10q22.1 | chr10; der(10) |
| RP11-632P2 | chr10:70330214-70506605 | 10q22.1 | chr10; der(10) |
| RP11-1106P13 | chr10:70506616-70653281 | 10q22.1 | chr10; der(10) |
| RP11-908J4 | chr10:70544654-70732390 | 10q22.1 | chr10; der(10) |
| RP11-671G17 | chr10:70821922-71027206 | 10q22.1 | chr10; der(3) |
| RP11-678L24 | chr10:71153343-71336334 | 10q22.1 | chr10; der(3) |
| RP11-643M21 | chr10:72224655-72410011 | 10q22.2 | chr10; der(3) |
| RP11-640K24 | chr10:73313882-73483445 | 10q22.2 | chr10; der(3) |
| RP11-1083I6 | chr10:74034423-74229058 | 10q22.2 | chr10; der(3) |
| RP11-668A2 | chr10:74850938-75042922 | 10q22.2 | chr10; der(3) |
| RP11-614P6 | chr10:75427422-75635499 | 10q22.2 | chr10; der(3) |
| RP11-642O17 | chr10:75827918-75994269 | 10q22.2-q22.3 | chr10; der(3) |
| RP11-816A22 | chr10:76633181-76807488 | 10q22.3 | chr10; der(3) |
| RP11-1006L21 | chr10:78676309-78892904 | 10q22.3 | chr10; der(3) |
| RP11-1130A7 | chr5:58400453-58572994 | 5q11.2 | chr5; der(5) |
| RP11-668D15 | chr5:58613049-58799717 | 5q11.2 | chr5; der(5) |
| RP11-719C2 | chr5:59122748-59288189 | 5q11.2 | chr5; der(5) |
| RP11-1082E13 | chr5:59232557-59409900 | 5q11.2 | chr5; weak der(5) |
| RP11-1110N23 | chr5:61540764-61717165 | 5q12.1 | chr5 |
| RP11-829M20 | chr5:67732992-67916370 | 5q13.1 | chr5 |
| RP11-846E14 | chr5:67730018-67956551 | 5q13.1 | chr5 |
| RP11-643L12 | chr5:67900262-68101923 | 5q13.1 | chr5; der(5) |
| RP11-1069B16 | chr5:68170119-68363482 | 5q13.1 | chr5; der(5) |
| Study | Deletion Size | GRCh38/hg38 Coordinates | Clinical Features |
|---|---|---|---|
| Cetin et al. 2013 | 887,687 | chr5:63,554,940-64,442,627 | height 3–10th centile, mild developmental delay, epilepsy, flat face, large forehead, depressed nasal bridge, deep-set eyes, low-set and dysplastic ears |
| Gnan et al. 2017 | 2,867,247 | chr5:64,286,356-67,153,603 | epilepsy, mild intellectual disability, behavioral abnormalities |
| Holder et. al 2015 | 4,045,298 | chr5:65,224,316-69,269,614 | height < 1st centile, hyperactivity, mild intellectual disability, large forehead, smooth philtrum |
| Jaillard et al. 2011 P2 | 5,756,904 | chr5:56,933,267-62,690,171 | mild developmental delay, flat feet, coarse face, large forehead, large nose, anteverted nostrils, long and proeminent philtrum, thin upper lip, hypotelorism, visual impairment, esotropia, hypermetropia, astigmatism, cryptorchidism, behavioural disorders |
| Lindstrand et al. 2013 P7 | 8,404,745 | chr5:53,332,485-61,737,230 | prominent nasal bridge and columella, posteriorly rotated ears, long palpebral fissures micrognathia, long fingers, underweight, late speech and psychomotor development, intellectual disability, autistic features, muscular hypotonia, hypermobility, myopia |
| Current study | 8,598,664 | chr5:59,356,735-67,955,399 | growth retardation, angioma of the right lower eyelid, ovarian cyst, polyhydramnios during pregnancy |
| Jaillard et al. 2011 P1 | 9,509,908 | chr5:60,058,538-69,568,446 | growth retardation, hydramnios during pregnancy, severe developmental delay, epilepsy, cardiac malformations, short arms, brachymesophalangy v, coarse face, flat face, large forehead, epicanthus, thin palpebral fissures, large nasal tip, flat nose, long philtrum, large mouth, thin upper lip, macroglossia, hypertelorism, visual impairment, esotropia, ptosis, hypermetropia, astigmatism, hair pigmentation irregularities, hirsutism, short neck, sacral dimple |
| Lindstrand et al. 2013 P6 | 10,181,207 | chr5:53,873,868-64,055,075 | prominent nasal bridge and columella, posteriorly rotated ears, long palpebral fissures, micrognathia severe overbite (requiring ramus osteotomy), long fingers and toes, marfanoid habitus, scoliosis (operated), atypical multiple nevi, underweight, low muscle mass |
| Jaillard et al. 2011 P3 | 15,356,299 | chr5:50,657,361-66,013,660 | growth retardation, severe developmental delay, post-axial hexadactyly (right foot), fingers joints laxity, spooned fingers, coarse face, flat face, frontal bossing, thin palpebral fissures, hypoplastic nares, bulbous nasal tip, prominent columella, short philtrum, thin upper lip, microretrognathia, visual impairment, esotropia, ptosis, sparse and thin hair, hypertrichosis, unstable walking |
| Jaillard et al. 2011 P4 | 17,269,546 | chr5:54,455,594-71,725,140 | growth retardation, severe developmental delay, increased nuchal translucency and short long bones during pregnancy, febrile seizures (normal eeg), cardiac malformations, short long bones and brachydactyly, coarse and flat face, prominent frontal forehead, large nose, brachycephaly, small ears, overfold pinnae, prominent cheeks, exotropia, short neck |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cellamare, A.; Coccaro, N.; Nuzzi, M.C.; Casieri, P.; Tampoia, M.; Maggiolini, F.A.M.; Gentile, M.; Ficarella, R.; Ponzi, E.; Conserva, M.R.; et al. Cytogenetic and Array-CGH Characterization of a Simple Case of Reciprocal t(3;10) Translocation Reveals a Hidden Deletion at 5q12. Genes 2021, 12, 877. https://doi.org/10.3390/genes12060877
Cellamare A, Coccaro N, Nuzzi MC, Casieri P, Tampoia M, Maggiolini FAM, Gentile M, Ficarella R, Ponzi E, Conserva MR, et al. Cytogenetic and Array-CGH Characterization of a Simple Case of Reciprocal t(3;10) Translocation Reveals a Hidden Deletion at 5q12. Genes. 2021; 12(6):877. https://doi.org/10.3390/genes12060877
Chicago/Turabian StyleCellamare, Angelo, Nicoletta Coccaro, Maria Cristina Nuzzi, Paola Casieri, Marilina Tampoia, Flavia Angela Maria Maggiolini, Mattia Gentile, Romina Ficarella, Emanuela Ponzi, Maria Rosa Conserva, and et al. 2021. "Cytogenetic and Array-CGH Characterization of a Simple Case of Reciprocal t(3;10) Translocation Reveals a Hidden Deletion at 5q12" Genes 12, no. 6: 877. https://doi.org/10.3390/genes12060877
APA StyleCellamare, A., Coccaro, N., Nuzzi, M. C., Casieri, P., Tampoia, M., Maggiolini, F. A. M., Gentile, M., Ficarella, R., Ponzi, E., Conserva, M. R., Cardarelli, L., Panarese, A., Antonacci, F., & Gesario, A. (2021). Cytogenetic and Array-CGH Characterization of a Simple Case of Reciprocal t(3;10) Translocation Reveals a Hidden Deletion at 5q12. Genes, 12(6), 877. https://doi.org/10.3390/genes12060877