
Generation and Genetic Correction of USH2A c.2299delG Mutation in Patient-Derived Induced Pluripotent Stem Cells

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
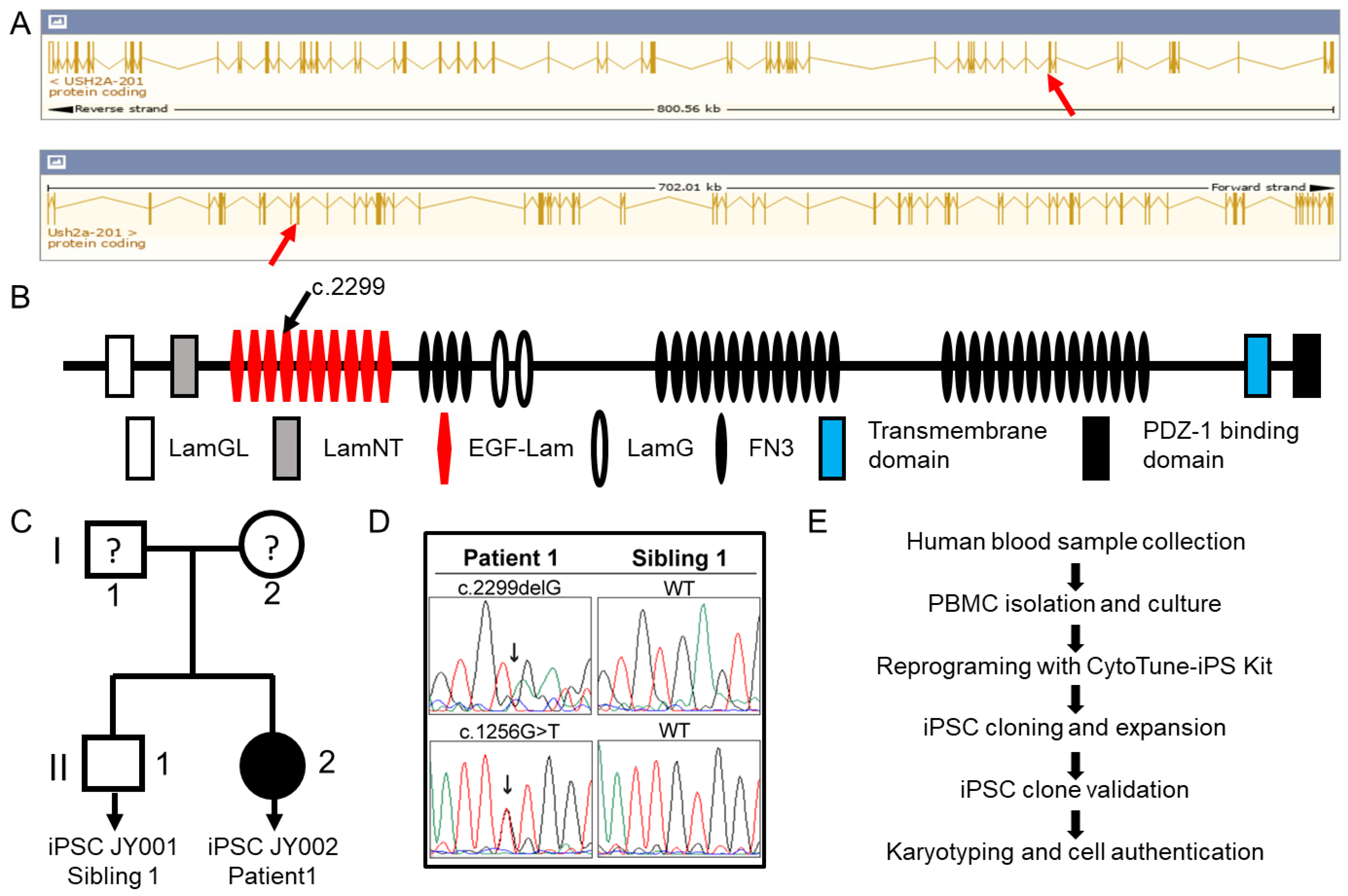
2.1. Patient Information
2.2. Generation of Patient-Derived Induced Pluripotent Stem Cells
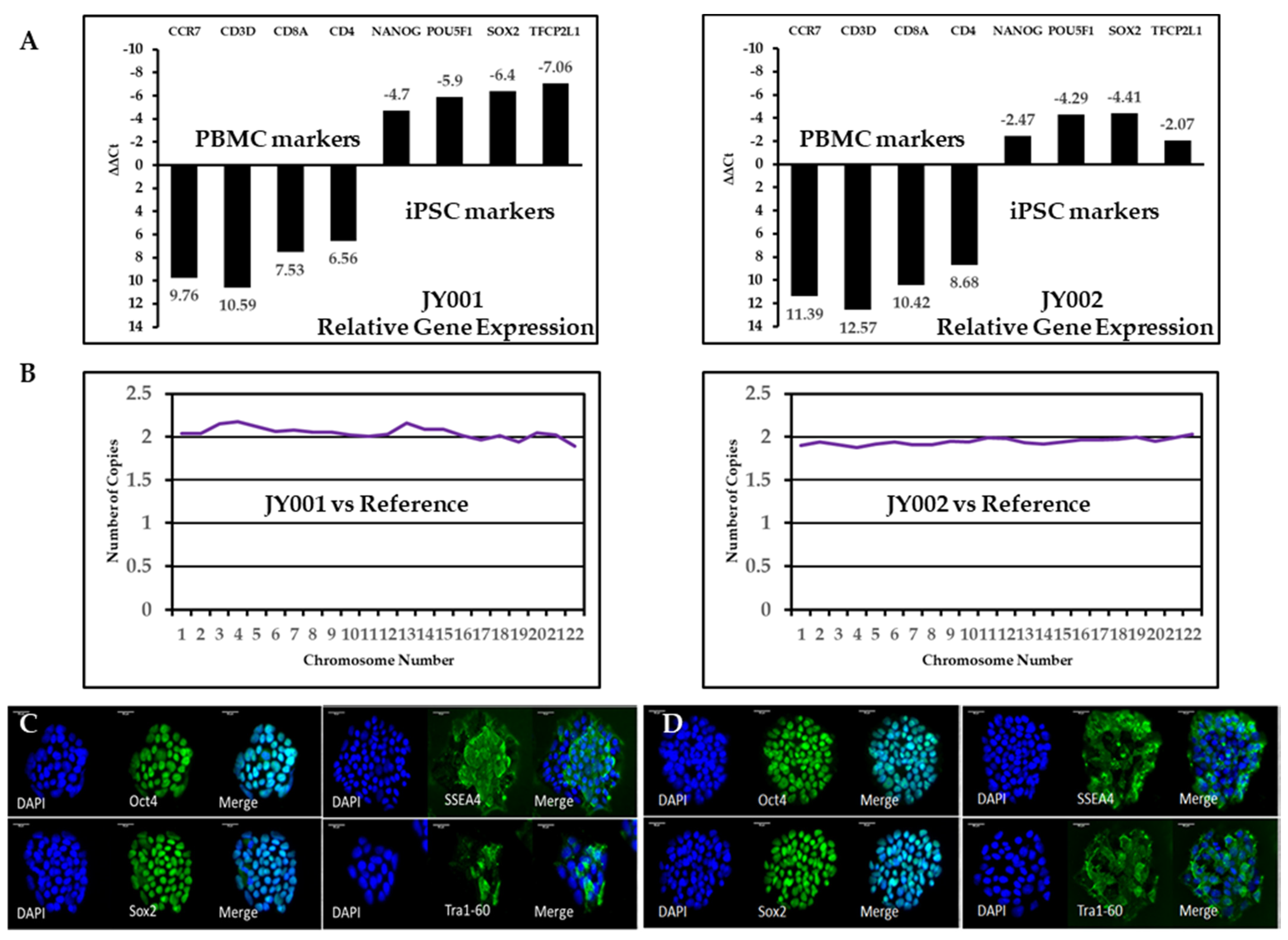
2.3. Reverse Transcription (RT) and Quantitative Polymerase Chain Reaction (qPCR) of Blood Cell Marker and Stem Cell Specific Genes
2.4. Immunostaining of Stem Cell Protein Markers and Trilineage Differentiation
2.5. Molecular Karyotype Using a Nanostring Molecular Probe Array
2.6. Human iPSC Line Authentication
2.7. Mycoplasma and Bacteria Detection
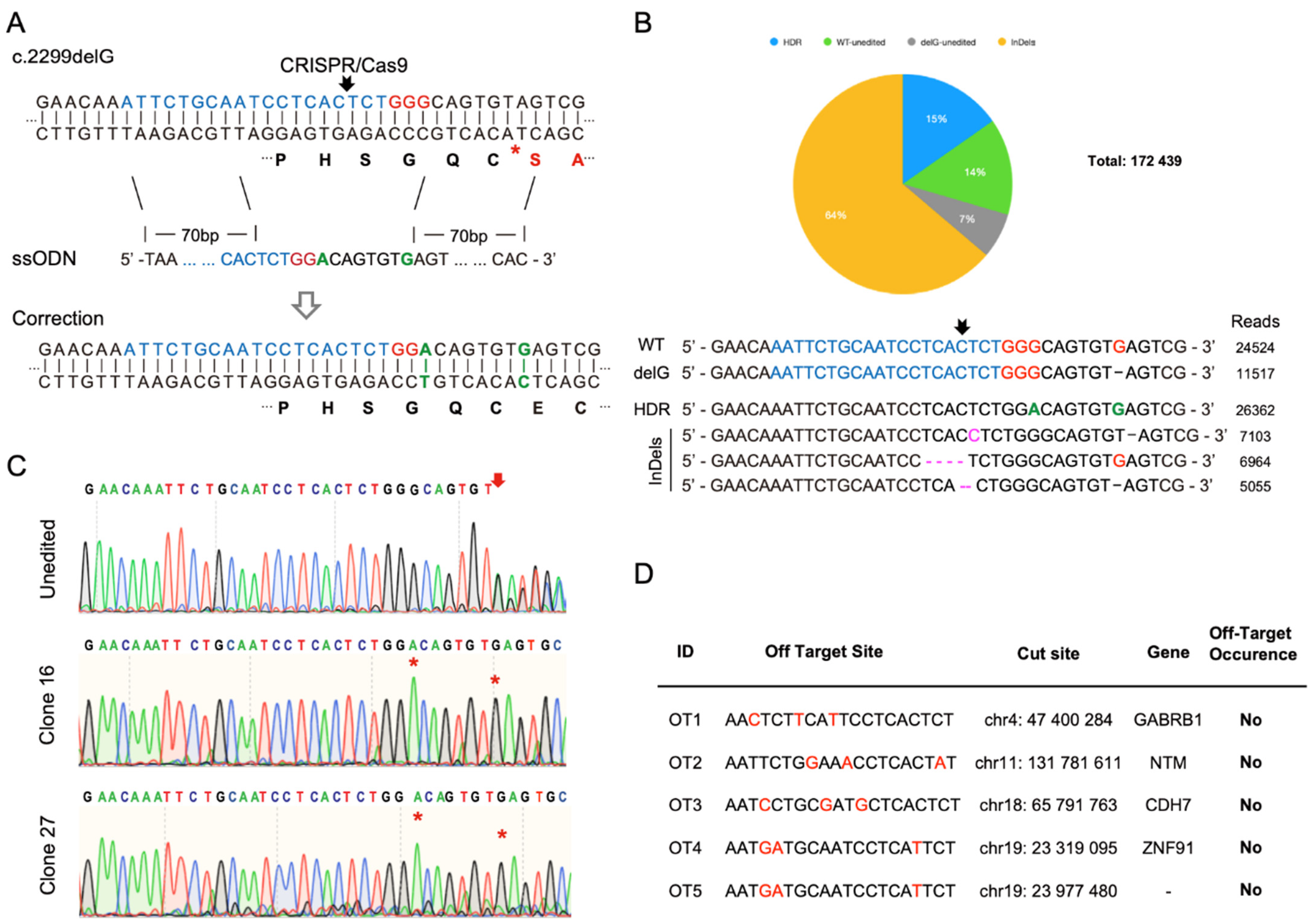
2.8. Single Guide RNA Design, CRISPR/Cas9 Treatment of Patient iPSCs, and Cell Cloning
2.9. Off-Target Analysis of CRISPR/Cas9 at Top Potential Genomic Loci
3. Results
3.1. Generation of Patient-Derived Induced Pluripotent Stem Cells
3.2. Validation of Generated Human iPSCs Using RT-qPCR and Immunostaining
3.3. Cell Molecular Karyotyping and Authentication
3.4. CRISPR/Cas9-Mediated Homology-Directed Repair and Single-Cell Cloning of Patient iPSCs
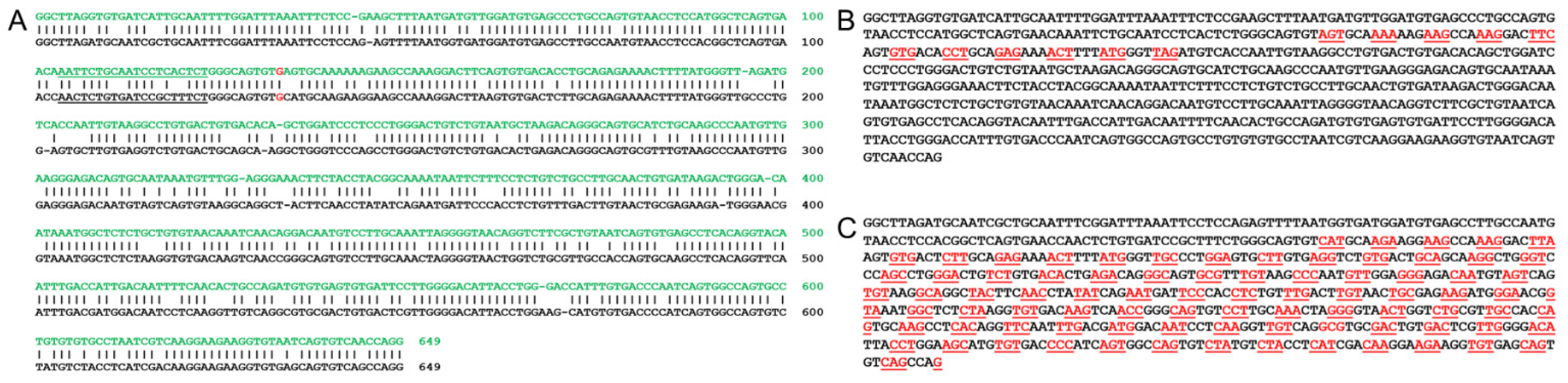
3.5. Bioinformatic Analysis of Utilizing a Mouse Model for a Human USH2A c.2299delG Study
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yan, D.; Liu, X.Z. Genetics and pathological mechanisms of Usher syndrome. J. Hum. Genet. 2010, 55, 327–335. [Google Scholar] [CrossRef]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Weston, M.D.; Eudy, J.D.; Fujita, S.; Yao, S.; Usami, S.; Cremers, C.; Greenberg, J.; Ramesar, R.; Martini, A.; Moller, C.; et al. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am. J. Hum. Genet. 2000, 66, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Pieke-Dahl, S.; van Aarem, A.; Dobin, A.; Cremers, C.W.; Kimberling, W.J. Genetic heterogeneity of Usher syndrome type II in a Dutch population. J. Med. Genet. 1996, 33, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Ouyang, X.; Patterson, D.M.; Du, L.L.; Jacobson, S.G.; Liu, X.Z. Mutation analysis in the long isoform of USH2A in American patients with Usher Syndrome type II. J. Hum. Genet. 2009, 54, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, B.; Tranebjaerg, L.; Brox, V.; Rosenberg, T.; Moller, C.; Beneyto, M.; Weston, M.D.; Kimberling, W.J.; Cremers, C.W.; Liu, X.Z.; et al. A common ancestral origin of the frequent and widespread 2299delG USH2A mutation. Am. J. Hum. Genet. 2001, 69, 228–234. [Google Scholar] [CrossRef]
- Ouyang, X.M.; Hejtmancik, J.F.; Jacobson, S.G.; Li, A.R.; Du, L.L.; Angeli, S.; Kaiser, M.; Balkany, T.; Liu, X.Z. Mutational spectrum in Usher syndrome type II. Clin. Genet. 2004, 65, 288–293. [Google Scholar] [CrossRef]
- Leroy, B.P.; Aragon-Martin, J.A.; Weston, M.D.; Bessant, D.A.; Willis, C.; Webster, A.R.; Bird, A.C.; Kimberling, W.J.; Payne, A.M.; Bhattacharya, S.S. Spectrum of mutations in USH2A in British patients with Usher syndrome type II. Exp. Eye Res. 2001, 72, 503–509. [Google Scholar] [CrossRef]
- Aller, E.; Najera, C.; Millan, J.M.; Oltra, J.S.; Perez-Garrigues, H.; Vilela, C.; Navea, A.; Beneyto, M. Genetic analysis of 2299delG and C759F mutations (USH2A) in patients with visual and/or auditory impairments. Eur. J. Hum. Genet. 2004, 12, 407–410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, X.Z.; Hope, C.; Liang, C.Y.; Zou, J.M.; Xu, L.R.; Cole, T.; Mueller, R.F.; Bundey, S.; Nance, W.; Steel, K.P.; et al. A mutation (2314delG) in the Usher syndrome type IIA gene: High prevalence and phenotypic variation. Am. J. Hum. Genet. 1999, 64, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Baux, D.; Blanchet, C.; Hamel, C.; Meunier, I.; Larrieu, L.; Faugere, V.; Vache, C.; Castorina, P.; Puech, B.; Bonneau, D.; et al. Enrichment of LOVD-USHbases with 152 USH2A genotypes defines an extensive mutational spectrum and highlights missense hotspots. Hum. Mutat. 2014, 35, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Le Quesne Stabej, P.; Saihan, Z.; Rangesh, N.; Steele-Stallard, H.B.; Ambrose, J.; Coffey, A.; Emmerson, J.; Haralambous, E.; Hughes, Y.; Steel, K.P.; et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 2012, 49, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Toualbi, L.; Toms, M.; Moosajee, M. USH2A-retinopathy: From genetics to therapeutics. Exp. Eye Res. 2020, 201, 108330. [Google Scholar] [CrossRef]
- Zou, B.; Mittal, R.; Grati, M.; Lu, Z.; Shu, Y.; Tao, Y.; Feng, Y.; Xie, D.; Kong, W.; Yang, S.; et al. The application of genome editing in studying hearing loss. Hear. Res. 2015, 327, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Gurumurthy, C.B.; Grati, M.; Ohtsuka, M.; Schilit, S.L.; Quadros, R.M.; Liu, X.Z. CRISPR: A versatile tool for both forward and reverse genetics research. Hum. Genet. 2016, 135, 971–976. [Google Scholar] [CrossRef]
- Mittal, R.; Nguyen, D.; Patel, A.P.; Debs, L.H.; Mittal, J.; Yan, D.; Eshraghi, A.A.; Van De Water, T.R.; Liu, X.Z. Recent Advancements in the Regeneration of Auditory Hair Cells and Hearing Restoration. Front. Mol. Neurosci. 2017, 10, 236. [Google Scholar] [CrossRef]
- Tang, Z.H.; Chen, J.R.; Zheng, J.; Shi, H.S.; Ding, J.; Qian, X.D.; Zhang, C.; Chen, J.L.; Wang, C.C.; Li, L.; et al. Genetic Correction of Induced Pluripotent Stem Cells From a Deaf Patient with MYO7A Mutation Results in Morphologic and Functional Recovery of the Derived Hair Cell-Like Cells. Stem Cells Transl. Med. 2016, 5, 561–571. [Google Scholar] [CrossRef]
- Chen, S.; Dong, C.; Wang, Q.; Zhong, Z.; Qi, Y.; Ke, X.; Liu, Y. Targeted Next-Generation Sequencing Successfully Detects Causative Genes in Chinese Patients with Hereditary Hearing Loss. Genet. Test. Mol. Biomark. 2016, 20, 660–665. [Google Scholar] [CrossRef]
- McLenachan, S.; Wong, E.Y.M.; Zhang, X.; Leith, F.; Moon, S.Y.; Zhang, D.; Chen, S.C.; Thompson, J.A.; McLaren, T.; Lamey, T.; et al. Generation of two induced pluripotent stem cell lines from a patient with compound heterozygous mutations in the USH2A gene. Stem Cell Res. 2019, 36, 101420. [Google Scholar] [CrossRef]
- Riera, M.; Patel, A.; Corcostegui, B.; Chang, S.; Corneo, B.; Sparrow, J.R.; Pomares, E. Generation of an induced pluripotent stem cell line (FRIMOi002-A) from a retinitis pigmentosa patient carrying compound heterozygous mutations in USH2A gene. Stem Cell Res. 2019, 35, 101386. [Google Scholar] [CrossRef]
- Sanjurjo-Soriano, C.; Erkilic, N.; Manes, G.; Dubois, G.; Hamel, C.P.; Meunier, I.; Kalatzis, V. Generation of a human iPSC line, INMi002-A, carrying the most prevalent USH2A variant associated with Usher syndrome type 2. Stem Cell Res. 2018, 33, 247–250. [Google Scholar] [CrossRef]
- Sanjurjo-Soriano, C.; Erkilic, N.; Manes, G.; Dubois, G.; Hamel, C.P.; Meunier, I.; Kalatzis, V. Generation of an iPSC line, INMi001-A, carrying the two most common USH2A mutations from a compound heterozygote with non-syndromic retinitis pigmentosa. Stem Cell Res. 2018, 33, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Mullins, R.F.; Streb, L.M.; Anfinson, K.; Eyestone, M.E.; Kaalberg, E.; Riker, M.J.; Drack, A.V.; Braun, T.A.; Stone, E.M. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. eLife 2013, 2, e00824. [Google Scholar] [CrossRef] [PubMed]
- Zaw, K.; Wong, E.Y.M.; Zhang, X.; Zhang, D.; Chen, S.C.; Thompson, J.A.; Lamey, T.; McLaren, T.; De Roach, J.N.; Wilton, S.D.; et al. Generation of three induced pluripotent stem cell lines from a patient with Usher syndrome caused by biallelic c.949C > A and c.1256G > T mutations in the USH2A gene. Stem Cell Res. 2020, 50, 102129. [Google Scholar] [CrossRef]
- Zurita-Díaz, F.; Ortuño-Costela, M.D.C.; Moreno-Izquierdo, A.; Galbis, L.; Millán, J.M.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Establishment of a human iPSC line, IISHDOi004-A, from a patient with Usher syndrome associated with the mutation c.2276G>T; p.Cys759Phe in the USH2A gene. Stem Cell Res. 2018, 31, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Erkilic, N.; Baux, D.; Mamaeva, D.; Hamel, C.P.; Meunier, I.; Roux, A.F.; Kalatzis, V. Genome Editing in Patient iPSCs Corrects the Most Prevalent USH2A Mutations and Reveals Intriguing Mutant mRNA Expression Profiles. Mol. Ther. Methods Clin. Dev. 2020, 17, 156–173. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, B.; Li, M.; Mo, F.; Mi, T.; Wu, Y.; Teng, Z.; Zhou, Q.; Li, W.; Hu, B. Precisely controlling endogenous protein dosage in hPSCs and derivatives to model FOXG1 syndrome. Nat. Commun. 2019, 10, 928. [Google Scholar] [CrossRef]
- Reiners, J.; Nagel-Wolfrum, K.; Jürgens, K.; Märker, T.; Wolfrum, U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006, 83, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yoshida, Y.; Yamanaka, S. Recent stem cell advances: Induced pluripotent stem cells for disease modeling and stem cell-based regeneration. Circulation 2010, 122, 80–87. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Spusta, S.C.; Mi, R.; Lassiter, R.N.; Stark, M.R.; Hoke, A.; Rao, M.S.; Zeng, X. Human neural crest stem cells derived from human ESCs and induced pluripotent stem cells: Induction, maintenance, and differentiation into functional schwann cells. Stem Cells Transl. Med. 2012, 1, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.R.; Tang, Z.H.; Zheng, J.; Shi, H.S.; Ding, J.; Qian, X.D.; Zhang, C.; Chen, J.L.; Wang, C.C.; Li, L.; et al. Effects of genetic correction on the differentiation of hair cell-like cells from iPSCs with MYO15A mutation. Cell Death Differ. 2016, 23, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Koehler, K.R.; Nie, J.; Longworth-Mills, E.; Liu, X.P.; Lee, J.; Holt, J.R.; Hashino, E. Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 583–589. [Google Scholar] [CrossRef]
- Chen, Y.C.; Tsai, C.L.; Wei, Y.H.; Wu, Y.T.; Hsu, W.T.; Lin, H.C.; Hsu, Y.C. ATOH1/RFX1/RFX3 transcription factors facilitate the differentiation and characterisation of inner ear hair cell-like cells from patient-specific induced pluripotent stem cells harbouring A8344G mutation of mitochondrial DNA. Cell Death Dis. 2018, 9, 437. [Google Scholar] [CrossRef]
- Jeong, M.; O’Reilly, M.; Kirkwood, N.K.; Al-Aama, J.; Lako, M.; Kros, C.J.; Armstrong, L. Generating inner ear organoids containing putative cochlear hair cells from human pluripotent stem cells. Cell Death Dis. 2018, 9, 922. [Google Scholar] [CrossRef] [PubMed]
- Muller, Q.; Beaudet, M.J.; De Serres-Berard, T.; Bellenfant, S.; Flacher, V.; Berthod, F. Development of an innervated tissue-engineered skin with human sensory neurons and Schwann cells differentiated from iPS cells. Acta Biomater. 2018, 82, 93–101. [Google Scholar] [CrossRef]
- Mattei, C.; Lim, R.; Drury, H.; Nasr, B.; Li, Z.; Tadros, M.A.; D’Abaco, G.M.; Stok, K.S.; Nayagam, B.A.; Dottori, M. Generation of Vestibular Tissue-Like Organoids From Human Pluripotent Stem Cells Using the Rotary Cell Culture System. Front. Cell Dev. Biol. 2019, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Boddy, S.L.; Romero-Guevara, R.; Ji, A.R.; Unger, C.; Corns, L.; Marcotti, W.; Rivolta, M.N. Generation of Otic Lineages from Integration-Free Human-Induced Pluripotent Stem Cells Reprogrammed by mRNAs. Stem Cells Int. 2020, 2020, 3692937. [Google Scholar] [CrossRef] [PubMed]
- Gosstola, N.C.; Huang, Z.; Tong, X.; Nourbakhsh, A.; Chen, Z.Y.; Dykxhoorn, D.M.; Zhong Liu, X. Characterization of UMi028-A-1 stem cell line that contains a CRISPR/Cas9 induced hearing loss-associated variant (V60L (c.178G > T)) in the P2RX2 gene. Stem Cell Res. 2020, 49, 102017. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Potter, J.; Kumar, S.; Ravinder, N.; Chesnut, J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017, 241, 136–146. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef]
- Besnard, T.; García-García, G.; Baux, D.; Vaché, C.; Faugère, V.; Larrieu, L.; Léonard, S.; Millan, J.M.; Malcolm, S.; Claustres, M.; et al. Experience of targeted Usher exome sequencing as a clinical test. Mol. Genet. Genomic Med. 2014, 2, 30–43. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EV | Cell No. | Cell Name | Locus Names | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D5S818 | D13S317 | D7S820 | D16S539 | VWA | TH01 | AM | TPOX | CSF1PO | |||
| JY001 | 11, 11 | 8, 12 | 10, 11 | 11, 13 | 15, 18 | 9.3, 9.3 | x, y | 11, 11 | 10, 12 | ||
| 0.78 (28/36) | CRL-2529 | CCD1124Sk | 11, 12 | 8, 12 | 10, 12 | 11, 13 | 15, 18 | 7, 9.3 | X, Y | 8, 11 | 10, 12 |
| 0.67 (24/36) | 557 | ROS-50 | 11, 11 | 8, 13 | 10, 11 | 8, 12 | 17, 18 | 6, 9.3 | X, Y | 11, 11 | 11, 12 |
| 0.67 (24/36) | 563 | HCC-78 | 11, 11 | 8, 12 | 9, 9 | 11, 13 | 15, 18 | 7, 9.3 | X, Y | 8, 11 | 11, 12 |
| 0.67 (24/36) | 740 | HD-MB03 | 11, 11 | 11, 12 | 9, 10 | 9, 11 | 18, 19 | 6, 9.3 | X, Y | 8, 11 | 10, 12 |
| 0.67 (24/36) | CRL-2096 | CCD-1076Sk | 11, 11 | 8, 11 | 10, 11 | 11, 13 | 16, 18 | 6, 9.3 | X, Y | 8, 8 | 11, 12 |
| 0.67 (24/36) | CRL-5882 | NCI-H1648[H1648] | 11, 11 | 12, 12 | 10, 11 | 11, 11 | 14, 17 | 7, 9.3 | X, Y | 8, 11 | 10, 12 |
| 0.67 (24/36) | CRL-5964 | NCI-BL2077 | 11, 11 | 8, 12 | 10, 12 | 12, 13 | 18, 20 | 8, 9.3 | X, Y | 8, 11 | 10, 11 |
| 0.67 (24/36) | CRL-7425 | Hs 688(A)T | 8, 11 | 12, 13 | 10, 11 | 9, 13 | 15, 18 | 7, 9 | X, Y | 8, 11 | 10, 12 |
| 0.67 (24/36) | CRL-7426 | Hs 688(B)T | 8, 11 | 12, 13 | 10, 11 | 9, 13 | 15, 18 | 7, 9 | X, Y | 8, 11 | 10, 12 |
| 0.67 (24/36) | CRL-7833 | Hs 172T | 11, 11 | 12, 12 | 8, 11 | 13, 13 | 18, 21 | 9.3, 9.3 | X, Y | 9, 11 | 11, 12 |
| EV | Cell No. | Cell Name | Locus Names | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D5S818 | D13S317 | D7S820 | D16S539 | VWA | TH01 | AM | TPOX | CSF1PO | |||
| JY002 | 11, 13 | 11, 12 | 10, 11 | 11, 12 | 14, 18 | 7, 9.3 | x, x | 8, 11 | 12, 12 | ||
| 0.78 (28/36) | 468 | SIG-M5 | 11, 13 | 11, 12 | 9, 9 | 11, 12 | 17, 19 | 7, 9.3 | X, X | 8, 11 | 12, 12 |
| 0.72 (26/36) | 42 | 697 | 11, 13 | 11, 12 | 10, 11 | 11, 12 | 16, 18 | 8, 9 | X, X | 8, 11 | 11, 12 |
| 0.72 (26/36) | 55 | A-498 | 11, 13 | 12, 12 | 10, 11 | 12, 12 | 16, 18 | 6, 9.3 | X, X | 8, 11 | 11, 12 |
| 0.72 (26/36) | 326 | SW-948 | 11, 11 | 10, 11 | 9, 11 | 11, 12 | 16, 18 | 6, 9.3 | X, X | 8, 11 | 12, 12 |
| 0.72 (26/36) | CCL237 | SW-948[SW-948] | 11, 11 | 10, 11 | 9, 11 | 11, 12 | 16, 18 | 6, 9.3 | X, X | 8, 11 | 12, 12 |
| 0.72 (26/36) | CRL-1594 | C-4I | 9, 11 | 11, 12 | 10, 11 | 11, 11 | 14, 14 | 9.3, 9.3 | X, X | 10, 11 | 12, 12 |
| 0.72 (26/36) | CRL-1595 | C-4II | 9, 11 | 11, 12 | 10, 11 | 11, 11 | 14, 14 | 9, 9.3 | X, X | 10, 11 | 12, 12 |
| 0.72 (26/36) | CRL-1718 | CCF-STTG1 | 12, 13 | 11, 13 | 10, 11 | 11, 12 | 17, 17 | 7, 8 | X, X | 8, 11 | 12, 12 |
| 0.72 (26/36) | CRL-7193 | Hs 228.T | 11, 12 | 8, 9 | 10, 11 | 11, 12 | 14, 18 | 8, 9.3 | X, X | 8, 11 | 11, 12 |
| 0.72 (26/36) | CRL-7242 | Hs 329.T | 11, 13 | 9, 11 | 11, 12 | 11, 12 | 17, 18 | 6, 9.3 | X, X | 8, 11 | 11, 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Lillywhite, J.; Zhu, W.; Huang, Z.; Clark, A.M.; Gosstola, N.; Maguire, C.T.; Dykxhoorn, D.; Chen, Z.-Y.; Yang, J. Generation and Genetic Correction of USH2A c.2299delG Mutation in Patient-Derived Induced Pluripotent Stem Cells. Genes 2021, 12, 805. https://doi.org/10.3390/genes12060805
Liu X, Lillywhite J, Zhu W, Huang Z, Clark AM, Gosstola N, Maguire CT, Dykxhoorn D, Chen Z-Y, Yang J. Generation and Genetic Correction of USH2A c.2299delG Mutation in Patient-Derived Induced Pluripotent Stem Cells. Genes. 2021; 12(6):805. https://doi.org/10.3390/genes12060805
Chicago/Turabian StyleLiu, Xuezhong, Justin Lillywhite, Wenliang Zhu, Zaohua Huang, Anna M Clark, Nicholas Gosstola, Colin T. Maguire, Derek Dykxhoorn, Zheng-Yi Chen, and Jun Yang. 2021. "Generation and Genetic Correction of USH2A c.2299delG Mutation in Patient-Derived Induced Pluripotent Stem Cells" Genes 12, no. 6: 805. https://doi.org/10.3390/genes12060805
APA StyleLiu, X., Lillywhite, J., Zhu, W., Huang, Z., Clark, A. M., Gosstola, N., Maguire, C. T., Dykxhoorn, D., Chen, Z.-Y., & Yang, J. (2021). Generation and Genetic Correction of USH2A c.2299delG Mutation in Patient-Derived Induced Pluripotent Stem Cells. Genes, 12(6), 805. https://doi.org/10.3390/genes12060805