Abstract
Chronic pancreatitis (CP) is a progressive, irreversible inflammatory disorder of the pancreas, which results from interrelations between different genetic and environmental factors. Genetic variants are the primary cause of the disease in early-onset nonalcoholic CP patients. Novel CP-associated genes are continuously emerging from genetic studies on CP cohorts, providing important clues for distinct mechanisms involved in CP development. On the basis of functional studies, the genetic alterations have been sub-grouped into CP-driving pathological pathways. This review focuses on the concept of CP as a complex disease driven by multiple genetic factors. We will discuss only well-defined genetic risk factors and distinct functional pathways involved in CP development, especially in the context of the early-onset nonalcoholic CP group. The diagnostic implications of the genetic testing will be addressed as well.
1. Introduction
Chronic pancreatitis (CP) is a long-lasting inflammatory disease starting from acute pancreatitis (AP) (sudden onset) through recurrent acute (more than one episode of acute pancreatitis) up to a progressive, irreversible inflammatory disorder. This disease is associated with periods of exacerbation and remission. It leads to irreversible pancreatic gland destruction with fibrosis. Consequently, permanent morphological changes in pancreas develop, followed by exocrine and endocrine malfunctioning [1,2]. Due to pancreas large functional reserves, exocrine and endocrine insufficiency occurs after about 90% of the gland’s parenchyma is destroyed by the inflammation process. Thus, long-term complications are observed after many years of the disease duration and they are mostly diagnosed in adult patients [1,2].
The prevalence of CP in adults is estimated at 27.4 per 100,000 people in Western Europe, whereas the prevalence of pediatric CP is 5.8 per 100,000 [3]. Causes of early-onset CP (below 35 years old) differ significantly from those described in older adults (alcohol abuse and smoking), and the most frequent are gene mutations, anatomical defects of pancreatic duct (pancreas divisum, ansa pancreatica), biliary tract diseases (choledocholithiasis, PSC), autoimmune pancreatitis, or lipid disorders (familial hypertriglyceridemia, hyperlipidemias, obesity) [1,4,5,6,7].
Although complex interrelations between environmental and genetic factors are surely involved in adult CP development, hereditary pancreatitis (HP) occurs approximately only in 4% of all CP cases in this group. On the contrary, the most significant role in CP development in early-onset pancreatitis, especially in children, is played by gene variants [4,5,6,8,9].
Genetic variants are important risk factors for early CP and can add specificity to the likely etiology, but they are neither necessary nor sufficient to make a diagnosis. Diagnosis of CP is based on studies of pancreas structure and function as a result of multidisciplinary cooperation of geneticist, gastroenterologist, radiologist and pathomorphologist. According to INSPIRE (International Study Group of Pediatric Pancreatitis: In Search for a Cure) group recommendations, the diagnosis of CP requires 1 of 3 criteria: (1) abdominal pain consistent with pancreatic origin, (2) evidence of exocrine pancreatic insufficiency and (3) evidence of endocrine-pancreatic insufficiency, which were associated with abnormalities on imaging studies (e.g., ductal changes (the irregular contour of the main pancreatic duct or its radicles, intra-ducting-filling defects, calculi, stricture or dilatation) and parenchymal tissues (generalized or focal enlargement, irregular contour cavities, calcifications, heterogeneous echotexture [10])). Imaging studies such as ultrasound, computer tomography (CT scans), magnetic resonance cholangiopacreatography (MRCP), endoscopic ultrasound (EUS) and endoscopic retrograde cholangiopancreatography (ERCP) are most reliable in diagnosing CP. MRCP provides the most accurate visualization of the pancreatic ductal system and has been regarded as the criterion standard for diagnosing chronic pancreatitis [2,11]. Exocrine pancreatic insufficiency (EPI) diagnosis can be made on the basis of several function tests, including fecal fat quantification (considered as the gold diagnostic standard for steatorrhea), fecal elastase-1 (FE-1) and 13C-mixed triglyceride breath test (13C-MTBT) [12]. At present, histopathology is not necessary to diagnose chronic pancreatitis [2,10,11].
HP develops mainly in childhood. According to current recommendations, the hereditary pancreatitis (HP) is recognized when pathogenic variants in PRSS1 gene (encoding cationic trypsinogen) are present or when acute pancreatitis or CP of unknown etiology occurred in 2 first-degree relatives or in 3 or more relatives of the second degree in two or more generations [5]. However, when the full criteria for HP family history are not met and the disease has been identified in at least one relative, the diagnosis of familial CP (FCP) is established [8].
Over the years, besides PRSS1, pathogenic variants in CFTR (encoding cystic fibrosis transmembrane conductance regulator gene) and SPINK1 (secretory trypsin inhibitor) genes were linked with increased risk of CP. Nevertheless, up to 30–40% of CP still remains idiopathic [5]. This raised the question of whether other genes are also involved. Finally, recent progress in molecular methodology made it possible to answer this question by identification of the additional genes, which variants confer the CP risk (e.g., CPA1, CTRC, TRPV6, CEL-HYB1 allele). Hence, currently, the CP is regarded as an oligogenic disease for which development depends on complex integrations between undefined number of genes and different functional pathways (Table 1, Figure 1). Moreover, according to some studies, complex genetic interactions are causally related to even more than 30% of recurrent acute and chronic pancreatitis [13].
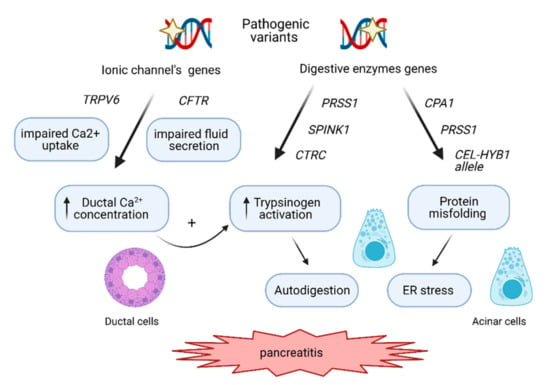
Figure 1.
The molecular pathways involved in CP development. The genetic risk in CP is mediated mainly by the trypsin-dependent pathway, where pathogenic variants in PRSS1 stimulate autoactivation of cationic trypsinogen within the acinar cells, leading to autodigestion of the pancreas. Pathogenic variants in SPINK1 and CTRC disrupt inhibition of cationic trypsinogen activation or its degradation respectively, also contributing to a higher level of trypsinogen activation. The variants in genes such as CFTR and TRPV6 impair ductal secretion and Ca2+ uptake, respectively, leading to increased Ca2+ concentration in the ductal fluid, which in turn stimulates (auto)activation of trypsinogen. The third mechanisms involve endoplasmic reticulum (ER) stress caused by genetic variants in CPA1, PRSS1 and by CEL-HYB1 alleles, inducing misfolding of these digestive enzymes and leading to acinar cell damage and inflammatory signaling.
The aim of this review is to present the current knowledge on the concept of CP as a complex disease caused by multiple genetic factors. We will discuss only well-defined genetic risk factors associated with nonalcoholic CP, with special attention to the early-onset disease group. The diagnostic implications of the genetic testing will be addressed as well.
2. The Molecular Pathways Involved in CP Development
2.1. The Trypsin-Dependent Pathway
The identification of activating pathogenic variants in cationic trypsinogen gene PRSS1 (a precursor of major pancreatic digestive enzyme) in hereditary pancreatitis supported the classical theory of pancreatitis as an autodigestive disease [14]. Pathogenic variants in PRSS1 stimulate autoactivation of cationic trypsinogen within the pancreas, triggering the activation of pancreatic digestive zymogenes cascade, which leads to autodigestion of the pancreas. Therefore, most of heterozygous pathogenic variants of PRSS1 are regarded as a cause of hereditary pancreatitis with autosomal dominant inheritance [15]. The genetic studies of different forms of this disease such as familial CP (FCP) and idiopathic CP (ICP) have identified pathogenic variants in other genes, including secretory trypsin inhibitor (SPINK1) and chymotrypisinogen C (CTRC) [16,17,18]. SPINK1 inhibitor is responsible for protecting the pancreas from premature, pathological trypsin activation, while CTRC is involved in trypsinogen degradation. Thus, pathogenic variants in SPINK1 and CTRC genes disrupt inhibition of cationic trypsinogen activation or its degradation, respectively [19].
2.2. The Misfolding-Dependent Pathway
Another pathway involved in CP genetic risk is endoplasmic reticulum (ER) stress. This mechanism is independent of the trypsin and is caused by misfolding of digestive enzymes due to genetic variants [20], which leads to diminished secretion, intracellular retention and endoplasmic reticulum stress. Consequently, acinar cell damage may occur as well as induction of inflammatory signaling.
Up to now, the best identified misfolding variants increasing CP risk or causing hereditary pancreatitis have been discovered in PRSS1 and CPA1 genes [21,22]. More recently, it was suggested that another genetic alteration: CEL and CELP hybrid allele (known as CEL-HYB1), may be associated with the misfolding-dependent pathway [23].
2.3. The Ductal Pathway
The third pathomechanism related to pancreatitis genetic risk is a ductal pathway which is caused by genetic variants in the genes encoding proteins involved in ductal secretion. So far, variants in two genes encoding transmembrane ionic channels: CFTR (cystic fibrosis transmembrane conductance regulator) and the TRPV6 (the transient receptor potential cation channel, subfamily V, member 6), seem to be involved. It is speculated that the variants of these genes cause the alteration of the ductal fluid Ca2+ concentration and, hence, increase ductal Ca2+ levels that eventually would stimulate autoactivation of trypsinogen [24,25].

Table 1.
Pathways associated with genetic variants involved in chronic pancreatitis development.
Table 1.
Pathways associated with genetic variants involved in chronic pancreatitis development.
| Gene (#OMIM) | Protein | CP Etiology | Genetic Variants # | Mechanisms | Pathway |
|---|---|---|---|---|---|
| PRSS1 (276000) | Cationic trypsinogen | HP, rarely sporadic CP | p.Asn29Ile, p.Ala16Val |
| Trypsin-dependent |
| p.Arg122His, p.Arg122Cys, p.Val39Ala |
| ||||
| Rare variants (e.g., Asp19Ala, p.Asp21Ala) |
| ||||
| Rare variants (e.g., p.Leu104Pro, p.Arg116Cys) |
| Misfolding-dependent | |||
| SPINK1 (167790) | pancreatic secretory trypsin inhibitor | ICP, | p.Asn34Ser |
| Trypsin-dependent |
| c.194+2T>C, and rare missense or nonsense variants |
| ||||
| CTRC (601405) | chymotrypisinogen C | ICP | p.Lys247_Arg254del, p.Arg254Trp, p.Val235Ile |
| Trypsin-dependent |
| CPA1 (114850) | carboxypeptidase A1 | HP and ICP | p.Asn256Lys, p.Ser282Pro, p.Arg382Trp |
| Misfolding-dependent |
| CEL-HYB1 (CEL: 114840) | Recombinant of carboxyl ester lipase and carboxyl ester lipase pseudogene | ICP | CEL-HYB1 allele (CEL and CELP recombinant) ((p.Thr488-Ile548 haplotype) |
| Misfolding-dependent |
| CFTR (602421) | cystic fibrosis transmembrane conductance regulator | ICP | p.Phe508del and other variants severely affecting CFTR expression and activity, p.Arg117His |
| Ductal |
| TRPV6 606680 | transient receptor potential cation channel, subfamily V, member 6 | ICP and familial CP | p.Glu575Lys, p.Arg345His, p.Arg483Trp |
| Ductal |
CP—chronic pancreatitis, HP—Hereditary pancreatitis, ICP—idiopathic chronic pancreatitis, familial CP, # examples of most frequent variants; full list of variants for PRSS1, SPINK1, CTRC, and CPA1 may be found in http://pancreasgenetics.org/ (accessed on 14 May 2020); CFTR and CEL variants—see references cited in the main text, TRPV6, according to Massamune et al. and Zou et al. [26,27].
3. The Genetic Variants Associated with CP
3.1. PRSS1
The most frequently found PRSS1 pathogenic variants belong to the trypsin-dependent pathway and cause hereditary pancreatitis with incomplete penetrance. The presence of these variants encourages cationic trypsinogen activity by different mechanisms, i.e., direct autoactivation stimulation, increase of CTRC-mediated processing of the activation peptide or by reduction of CTRC-dependent degradation [25]. The most common PRSS1 variants worldwide are p.Arg122His and p.Asn29Ile, which are present in 60–80% of HP cases [6,28,29]. The PRSS1 pathogenic variants such as p.Arg112Cys, p.Ala16Val and p.Glu79Lys are less frequent and p.Asp19Ala, p.Asp21Ala, p.Asp22Gly and p.Lys23Arg are rare variants [6,30]. The distributions of these PRSS1 variants may differ between different countries. For example, in Polish children with HP (age at onset around 8.5 years), the most common pathogenic variant is p.Arg122His (34%), which is consistent with the other data [28,31,32]. However, the second most prevalent pathogenic variant is p.Arg122Cys (27%), for which frequency is much lower in other cohorts and ranges from 0% to 1.5% [28,31,32,33]. The p.Asn29Ile pathogenic variant, the second most frequent in the world, in the Polish patients was observed in only 12% of the cases, and the very rare p.Glu79Lys pathogenic variant worldwide was found in as many as 7% of the cases.
Functionally, the p.Arg122Cys and p.Arg122His variants prevent CTRC-dependent trypsinogen degradation, whereas the p.Asn29Ile affects trypsinogen biochemistry, strengthening the process of its autoactivation [34]. Similarly, in case of variant p.Ala16Val, autoactivation is enhanced. In this case, the activation peptide of trypsinogen to CTRC-mediated processing is more sensitive [34,35]. However, the p.Ala16Val variant shows reduced disease penetrance compared to p.Arg122His, which may be explained by lower trypsin levels generated by the p.Ala16Val variant.
Rare PRSS1 variants (p.Asp19Ala, p.Asp21Ala, p.Asp22Gly, p.Lys23Arg and p.Lys23_Ile24insIleAspLys) have an impact on the cationic trypsinogen activation peptide and hence stimulate autoactivation in a CTRC-independent way [36,37].
The distinctive mechanism based on both increased trypsinogen activation and secretion was observed in case of the PRSS1-PRSS3P2 conversion allele found in two unrelated patients in Poland and Germany [38]. The converted region between exon 3 of the PRSS1 and PRSS3P2 contained three non-synonymous amino acid changes, p.Ser115Thr, p.Arg116Pro and p.Arg122His. The pathogenic effect of the PRSS1-PRSS3P2 conversion allele is due to increased trypsinogen activation and secretion, attributed to p.Arg122His and p.Arg116Pro variants, respectively.
The PRSS1 variants associated with the misfolding-dependent pathway are rare. They cause reduced secretion, intracellular retention and elevated ER stress markers. The p.Leu104Pro and p.Arg116Cys were detected in HP with incomplete penetrance, whereas p.Cys139Phe, p.Cys139Ser and p.Gly208Ala are mostly associated with sporadic CP [20,22].
The PRSS1 variants presumed to cause CP, such as p.Asp100His, p.Cys139Phe, and p.Lys92Asn, p.Ser124Phe and p.Gly208Ala, lead to diminished trypsinogen secretion in in vitro studies, indicating the misfolding-dependent pathway as a probable pathomechanism in this case [39].
3.2. SPINK1
There are two major molecular mechanisms conferring the SPINK1-related elevated risk of chronic pancreatitis. Both of them, reduced expression of mRNA and protein, cause diminished production of protective trypsin inhibitor [16].
The most frequent variant found in SPINK1 is p.Asn34Ser, which according to meta-analysis studies was detected in 9.7% of CP alleles (469/4842) compared to 1% of control alleles (96/9714) [40]. In Polish early-onset nonalcoholic CP patients, variants in SPINK1 have a frequency of 8–28% [6,8,41]. In European populations, the p.Asn34Ser increases CP risk about 10–15-fold [42]. The exact mechanism of p.Asn34Ser molecular pathophysiology remains unclear. According to recent studies by Szabó et al., p.Asn34Ser variant does not impact trypsin inhibition or enhanced SPINK1 degradation [43]. Moreover, only 1% of the p.Asn34Ser carriers develop symptoms of acute or chronic pancreatitis, indicating that other risk factors are involved [44,45]. The haplotype context of p.Asn34Ser was hypothesized to be one of them [43].
Moreover, it has not been established yet whether heterozygous p.Asn34Ser, when present together with additional genetic or environmental risk factors, should be treated as a disease-modifying or causative factor [43]. It should be noted, however, that no p.Asn34Ser homozygote has ever been found in a healthy population and patients with homozygous p.Asn34Ser develop severe symptoms of CP [18,46]. Therefore, it is assumed that SPINK1 p.Asn34Ser heterozygous variants contribute to CP, whereas p.Asn34Ser homozygous are causative of CP [47,48].
Unlike in the case of p.Asn34Ser, the pathomechanism of another frequent SPINK1 variant—the intronic c.194+2T>C—is rather clear as it leads to skipping of exon 3 [16,49,50]. Besides, around 60, mainly rare or private, variants in SPINK1 have been reported, according to the HGMD database. Although in case of certain variants miscellaneous mechanisms were postulated to affect SPINK1 activity, the reduced expression and/or secretion seem to be mostly involved [16].
3.3. CTRC
The pathogenic variants of CTRC generally cause loss of protein function [51,52]. For example, the p.Lys247_Arg254del variant makes the protein inactive and easily degraded, and the p.Arg254Trp leads to degradation by trypsin [51]. In adult CP patients, the p.Lys247_Arg254del and the p.Arg254Trp are rare, found with frequency of 0.1% to 1.5% and 1.2% to 2%, respectively (and with 5-fold increased CP risk on average) [17,18,53]. In early-onset (pediatric) CP patients, however, the p.Lys247_Arg254del (5.3%) and p.Arg254Trp (4.6%) variants are relatively frequent (combined frequency of 9.6%), as demonstrated in Polish CP children [54]. In this cohort, p.Arg254Trp and p.Lys247_Arg254del are associated with 19-fold and 5-fold increased CP risk compared to controls, respectively.
There is a frequent p.Gly60Gly CTRC heterozygous variant found in 30–40% of CP patients, including early-onset CP patients [17,54,55]. In the early-onset patients, CP risk is increased by 2.5-fold in case of p.Gly60Gly heterozygosity and by 24-fold in p.Gly60Gly homozygosity [54]. The mechanism associated with p.Gly60Gly is unclear, however, some data may suggest reduced CTRC mRNA expression, possibly caused by altered pre-mRNA splicing [30].
3.4. CPA1
Digestive carboxypeptidases are pancreatic metalloproteases, which hydrolyze C-terminal peptide bonds in dietary polypeptide chains. In 2013, Witt et al. reported that variants in the CPA1 (carboxypeptidase A1) gene are strongly associated with CP (OR ~ 25), and, in particular, with sporadic cases of CP with early onset (OR ~ 80) [21]. It was shown that CPA1 variants confer increased CP risk by misfolding affecting secretion, causing intracellular retention and inducing ER stress, rather than affecting trypsin activity. Nevertheless, it has not been analyzed if CPA1 variants are causative risk factors for the familial or hereditary disease. However, a study by Kujko et al. shed light on this issue, showing that the novel CPA1 variant p.Ser282Pro co-segregated with pancreatitis in two Polish families with autosomal dominant hereditary pancreatitis [56]. The age of onset for index patients in these two families was 17 and 12 years. The p.Ser282Pro CPA1 variant causes protein retention and degradation (indicative for misfolding), leading to ER stress. Interestingly, the same mechanism has also previously been described for the p.Leu104Pro PRSS1 variant [57].
Importantly, recently, a study on a mouse model for the misfolding-dependent pathway was published. The authors constructed the knock-in mice harboring the most frequent CPA1 human variant: p.Asn256Lys, and demonstrated development of spontaneous and progressive CP together with ER stress symptoms in their pancreas [58].
3.5. CEL-HYB1 Allele
The CEL gene encodes a carboxyl ester lipase (previously known as bile salt-stimulated lipase), which is a digestive enzyme produced in pancreas acinar cells and secreted into the small intestine. The CEL protein is activated by bile salts and is involved in hydrolysis and absorption of cholesterol and lipid-soluble vitamin esters [59].
In 2015, Fjeld et al. characterized a hybrid allele CEL-HYB (currently referred to as CEL-HYB1), which originates from a non-allelic homologous recombination between CEL and its pseudogene CELP. The average population frequency of CEL-HYB1 recombinant was 0.5–1%, while in a group of German and French idiopathic CP, it was 5-fold overrepresented [23]. The cell culture experiments revealed intracellular retention and poor secretion of the hybrid protein. It was proposed that the CEL-HYB1 allele may increase CP risk through the misfolding-dependent pathway [23]. However, the study of Zou et al. showed that the CEL-HYB1 allele is not present in three independent CP Asian cohorts—from China, Japan and India. Hence, the authors came up with an idea that the CEL-HYB1 allele could be a European ethnicities-specific CP risk factor [60]. Unexpectedly, another European replication study on Polish CP pediatric cohorts failed to find any significant difference in the CEL-HYB1 allele frequency between CP patients and controls (4.8% vs. 2.4%, p = 0.16). Moreover, the distribution of alleles in the Polish control group was higher than in German, French and Norwegian controls: 2.4% (12/500) vs. 0.7–1%, 0.7% and 0.3%, respectively [22,61,62].
In 2020, further studies with German and French patients revealed the presence of two amino acid substitutions in CEL-HYB, p.Thr488-Ile548, in CP cases but not in controls. On the contrary, the frequency of another, similar variant, thep.Thr488-Thr548, was not significantly different between patients and control groups. It was suggested that the CEL-HYB1 allele harboring the p.Thr488-Ile548 haplotype may be responsible for increased risk of CP. Surprisingly, the functional assays on p.Thr488-Ile548 and p.Thr488-Thr548 variants revealed that both variants show similar pathogenicity and cause proteotoxic protein-misfolding [63]. The discordance between genetic and functional data in case of the p.Thr488-Thr548 haplotype indicates that the role of CEL-HYB1 allele variants in CP development remains unclear and requires further investigation [63].
3.6. CFTR
The CFTR gene encodes the cystic fibrosis transmembrane conductance regulator, the ABC family membrane transport protein channel, localizing in the apical plasma membrane of epithelial cells [64].
Pathogenic variants in the CFTR gene are responsible for cystic fibrosis but are also risk factors of CFTR-related disorders. CF patients may clinically present with exocrine pancreatic insufficiency and, rarely, pancreatitis. Studies on cystic fibrosis indicate, however, that not all CFTR variants are correlated with pancreas insufficiency [65,66,67].
CFTR is crucial for the maintenance of proper ductal fluid levels and consistency and plays a role in regulating bicarbonate (HCO3−) secretion [68]. Thus, loss of CFTR function may cause decreased bicarbonate secretion and impaired ductal flushing [24,69]. It has been suggested that the decrease in bicarbonate secretion may prolong the time of zymogens transport to the duct and decrease pH, and thereby promote the autoactivation of trypsinogen. Activated trypsin would further inhibit CFTR channel activity and bicarbonate secretion, leading to intraductal acidosis. Acidosis would further enhance trypsinogen autoactivation, which could induce development of the pancreatitis [69]. This mechanism, however, may be particularly important in HP, where the mutated trypsinogens (i.e., with the p.Arg122His in PRSS1) are easily autoactivated.
Mutations in the CFTR gene are divided into six classes, depending on the severity of their impact on the protein synthesis and activity [70]. In general, compound heterozygous state for two severe mutations leads to cystic fibrosis, while when at least one allele harbors the mild CFTR allele, the CFTR-related disorder occurs. Hence, being a compound heterozygote for one severe and one mild CFTR mutation is considered as a strong risk or even causative factor for chronic pancreatitis [71]. Moreover, it has also been proven that p.Phe508del heterozygosity confers a 2.5 times increased risk for CP, while the mild p.Arg117His variant increases risk by about 4-fold [47]. There are over 2000 variants in the CFTR gene and involvement in pancreatitis or other CFTR-related disorders of several of them remains controversial [71]. For example, it has been postulated that certain non-CF-causing CFTR variants (e.g., p.Arg75Gln, p.Leu997Phe) leading to a selective, bicarbonate transportation defect in CFTR channel function may be risk factors for chronic pancreatitis [72]. While functional measurements seem to support this finding, a genetic association between these variants and chronic pancreatitis cannot be confirmed [47,53].
3.7. TRPV6
TRPV6 is a tetrameric, calcium-selective channel participating in epithelial Ca2+ (re)absorption [73]. The TRPV6 is predominantly expressed in ductal cells, which suggests a role in controlling calcium concentration of pancreatic juice, however its precise function is not yet fully discovered [74].
In 2020, two research groups showed that loss of function variants of TRPV6 are associated with risk of chronic pancreatitis in patients from Japan, Germany, France and China [26,27]. During our research towards identification of novel CP susceptibility genes using a whole exome sequencing approach, we also found overrepresentation of the novel defective TRPV6 variants in Polish CP pediatric patients (data unpublished, [75]). These findings taken together indicate that TRPV6 is a novel CP susceptibility gene.
In nonalcoholic early-onset CP, TRPV6 variants contribute to disease pathogenesis in 1.5–4% of the patients [26,27]. Since it is speculated that TRPV6 plays a role in Ca2+ removal from the ducts, the loss-of-function variants could cause elevated ductal Ca2+ concentration, which would stimulate autoactivation of trypsinogen and increased trypsin activity. However, the exact mechanism of how TRPV6 variants alter pancreatic Ca2+ balance requires further investigation. Probably, the cumulative genetic handicap is necessary to develop CP, which is supported by the fact that TRPV6 variants often coexist with pathogenic variants in other CP susceptibility genes such as SPINK1 and CFTR, according to Masamune et al. [26].
4. Complex Genetic Interactions
It is known that CP may develop due to simultaneous presence of variants in different recessive genes [76]. The paradigm of gene–gene interactions appears to be a co-existence of a variant increasing the recurrent trypsin activation risk (e.g., in PRSS1, CFTR genes), together with a variant protecting the pancreas from active trypsin (e.g., SPINK1, CTRC genes) or chronic inflammation. Such interactions are well-documented in patients who carry both SPINK1 and CFTR variations. Pancreatitis risk increases approximately 60-fold by having the heterozygote genotype in SPINK1 and CFTR (one variant in one allele of SPINK1 and one variant in one allele of CFTR), whereas the CP risk increases up to 900-fold by having a CFTR compound heterozygote genotype (two heterogeneous variants) and the SPINK1 p.Asn34Ser variant in one allele [67,77]. Coinheritance of the SPINK1 p.Asn34Ser in one allele and at least one abnormal allele in the CFTR accounts for 1.5–7.7% of the cases, depending on the population analyzed. Trans-heterozygotes of SPINK1 p.Asn34Ser and CTRC (defect in one allele of SPINK1 and one allele of CTRC) were detected in 1% of German CP patients and in 2.3% of young French ICP patients [17,18].
While the CFTR/SPINK1 trans-heterozygosity has a synergistic effect [72], the SPINK1/CTRC trans-heterozygosity is rather connected with an additive or a multiplicative impact, as those genes are involved in the same pathway [78]. Similarly, trans-heterozygosity of TRPV6 and CFTR variants may have an additive effect (two genes belong to the ductal pathway), whereas in case of TRPV6/SPINK1, trans-heterozygosity may show a synergistic effect [24].
5. Diagnostic Implications
According to the European guidelines, all patients with a family history or early-onset disease should be offered genetic testing for associated genes [2]. Since alcohol abuse is the predominant cause of the disease in up to 70% of adult cases, genetic screening for every CP patient is not recommended. In pediatric cases, the genetic screening should be offered for the patients with a second episode of idiopathic AP or first episode of idiopathic AP, and a family history of AP or CP. In these patients, the analysis of the presence of the pathogenic variants in PRSS1, CPA1, SPINK1, CTRC and CFTR genes as well as testing for the CEL gene pathogenic hybrid allele is recommended [11]. Hereditary pancreatitis associated with mutations in PRSS1, especially p.Arg122His, could considerably increase the risk of pancreatic adenocarcinoma (87 times more likely at age 55) [79]. Since more than 95% of pancreatic cancers are diagnosed at a later stage, the 5-year survival rate remains only 6% [80]. Therefore, in Poland, the USA and many other countries, pancreatic cancer screening (neoplasm markers, EUS and/or ultrasound/yearly) is recommended for all CP patients ≥ 35 years old who carry pathogenic variants in PRSS1 [81,82].
In recent years, the knowledge about molecular aspects of chronic pancreatitis has markedly increased, and therefore, the current guidelines need further updates. In this respect, the genetic screening of the TRPV6 gene should be included for early-onset CP patients. Besides, in the light of the novel data, screening towards functionally defective CEL-HYB1 allele variants seems to be more appropriate than investigating the presence of the allele itself. Moreover, there are still several questions unanswered with the respect to the multifactorial etiology of idiopathic CP. Here, both molecular diagnostic and genetic counseling are becoming more important and require further consideration.
In case of probands’ relatives, molecular testing is a basis for establishing a carrier status, which enables not only CP risk assessment, but also implementation of preventive actions or, in milder cases, even early diagnosis [83,84]. While in autosomal dominant HP due to PRSS1 or CPA1 pathogenic variants the case is rather clear, it becomes much more complicated in idiopathic CP when other genes are involved. The combinations of genetic factors may be epistatic, while others are additive, therefore the genetic risk assessment should be handled on a case-by-case basis. Unfortunately, no systematic approach is available to predict the effects of most of these complex genotypes [13]. Since it is widely accepted that the effect of the variant in one CP-connected gene is affected by the presence of a variant in another gene, the genetic risk assessment becomes challenging. Since no systematic approach has been elaborated, in case of the complex genotypes, individual assessment for each person is needed [13].
In 2013, Masson et al. proposed a clinically useful classification of variants and genotypes found in CP patients, which were divided into three categories: causative, contributory or neutral. The factors considered in this assessment include: allelic frequency in healthy and CP groups, functional defects, the relative importance of their associated genes in the pathogenesis of chronic pancreatitis and intergenic interactions, when applicable [47].
More recently, another group proposed a scoring system, which enables the classification of variants into four categories: (1) established risk alleles, (2) likely risk alleles, (3) uncertain risk alleles and (4) benign variants. This categorization of variants is based on the results of in vitro experiments, functional and epidemiological data and in silico predictions. Importantly, the authors elaborated a very clear scoring system, which enables rather unequivocal classification. This scoring system was used to evaluate the status of all variants known at the time in SPINK1 (n = 56) and CTRC (n = 87). In the case of SPINK1, 25/56 variants were scored as established risk alleles, 8/56 as likely risk alleles, 17/56 as uncertain and 6/56 as benign. In the case of CTRC, the quantity of individual groups was: 24/87, 4/87, 22/87 and 37/87, respectively [84].
These results, otherwise very important and useful in clinical settings, also illustrate the complexity of interpretational issues in CP genetics, which still involves not only establishing of individual variant status, but also assessment of intergenic relations.
6. Conclusions
Nonalcoholic chronic pancreatitis is a complex disease which may be caused by multiple genetic factors. Genetic testing for CP patients with unknown etiology is recommended by analyzing major CP-associated genes such as PRSS1, CPA1, CTRC, CFTR, SPINK1 and TRPV6, especially in early-onset cases and after the other major causes of the disease have been excluded. The diagnostic utility of the CEL-HYB1 allele and its variants requires further research.
Genetic counseling both before and after molecular testing is strongly recommended in all patients. Importantly, CP patients having genetic variants should be carefully followed by the clinicians since they may have a high risk of developing further medical complications such as pancreatic exocrine insufficiency, diabetes mellitus and pancreatic cancer [85].
The progress and availability of large-scale molecular methods enable unprecedented insight into hereditary pancreatitis genetics. Hence, proof that variants in other genes contribute to early-onset nonalcoholic chronic pancreatitis will surely emerge. Clearly, the genetic guidelines for clinical use are needed to help to stratify the risk of CP associated with different genetic factors.
Author Contributions
Conceptualization A.M.R. and K.W.-T.; Writing—Original Draft Preparation, A.M.R.; Writing—Review and Editing, K.W.-T. and G.O.; Visualization, A.M.R.; Project Administration, A.M.R.; Funding Acquisition, A.M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NCN, grant number: 2015/19/B/NZ5/02224.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The sponsors had no role in the design, execution, interpretation, or writing of the study.
References
- Braganza, J.M.; Lee, S.H.; McCloy, R.F.; McMahon, M.J. Chronic pancreatitis. Lancet 2011, 377, 1184–1197. [Google Scholar] [CrossRef]
- Löhr, J.M.; Dominguez-Munoz, E.; Rosendahl, J.; Besselink, M.; Mayerle, J.; Lerch, M.M.; Haas, S.; Akisik, F.; Kartalis, N.; Iglesias-Garcia, J.; et al. United European Gastroenterology evidence-based guidelines for the diagnosis and therapy of chronic pancreatitis (HaPanEU). United Eur. Gastroenterol J. 2017, 5, 153–199. [Google Scholar] [CrossRef] [PubMed]
- Sellers, Z.M.; MacIsaac, D.; Yu, H.; Dehghan, M.; Zhang, K.Y.; Bensen, R.; Wong, J.J.; Kin, C.; Park, K.T. Nationwide Trends in Acute and Chronic Pancreatitis Among Privately Insured Children and Non-Elderly Adults in the United States, 2007–2014. Gastroenterology 2018, 155, 469–478.e461. [Google Scholar] [CrossRef]
- Werlin, S.L.; Kugathasan, S.; Frautschy, B.C. Pancreatitis in children. J. Pediatr. Gastroenterol. Nutr. 2003, 37, 591–595. [Google Scholar] [CrossRef]
- Nydegger, A.; Couper, R.T.; Oliver, M.R. Childhood pancreatitis. J. Gastroenterol. Hepatol. 2006, 21, 499–509. [Google Scholar] [CrossRef]
- Wejnarska, K.; Kolodziejczyk, E.; Wertheim-Tysarowska, K.; Dadalski, M.; Sobczynska-Tomaszewska, A.; Kierkus, J.; Bal, J.; Rygiel, A.M.; Oracz, G. The Etiology and Clinical Course of Chronic Pancreatitis in Children with Early Onset of the Disease. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Löhr, J.M.; Beuers, U.; Vujasinovic, M.; Alvaro, D.; Frøkjær, J.B.; Buttgereit, F.; Capurso, G.; Culver, E.L.; de-Madaria, E.; Della-Torre, E.; et al. European Guideline on IgG4-related digestive disease—UEG and SGF evidence-based recommendations. United Eur. Gastroenterol. J. 2020, 8, 637–666. [Google Scholar] [CrossRef] [PubMed]
- Sobczyńska-Tomaszewska, A.; Bak, D.; Oralewska, B.; Oracz, G.; Norek, A.; Czerska, K.; Mazurczak, T.; Teisseyre, M.; Socha, J.; Zagulski, M.; et al. Analysis of CFTR, SPINK1, PRSS1 and AAT mutations in children with acute or chronic pancreatitis. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 299–306. [Google Scholar] [CrossRef]
- Oracz, G.; Kolodziejczyk, E.; Sobczynska-Tomaszewska, A.; Wejnarska, K.; Dadalski, M.; Grabarczyk, A.M.; Kierkus, J.; Woynarowski, M.; Wertheim-Tysarowska, K.; Ryzko, J.; et al. The clinical course of hereditary pancreatitis in children—A comprehensive analysis of 41 cases. Pancreatology 2016, 16, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Morinville, V.D.; Husain, S.Z.; Bai, H.; Barth, B.; Alhosh, R.; Durie, P.R.; Freedman, S.D.; Himes, R.; Lowe, M.E.; Pohl, J.; et al. Definitions of pediatric pancreatitis and survey of present clinical practices. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 261–265. [Google Scholar] [CrossRef]
- Párniczky, A.; Abu-El-Haija, M.; Husain, S.; Lowe, M.; Oracz, G.; Sahin-Tóth, M.; Szabó, F.K.; Uc, A.; Wilschanski, M.; Witt, H.; et al. EPC/HPSG evidence-based guidelines for the management of pediatric pancreatitis. Pancreatology 2018, 18, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Wejnarska, K.; Kołodziejczyk, E.; Ryżko, J.; Oracz, G. Comparison of 72-hour fecal fat quantification and the 13C-mixed triglyceride breath test in assessing pancreatic exocrine sufficiency in children with chronic pancreatitis. Dev. Period Med. 2016, 20, 222–227. [Google Scholar] [PubMed]
- Shelton, C.; LaRusch, J.; Whitcomb, D.C. Pancreatitis Overview; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Whitcomb, D.C.; Gorry, M.C.; Preston, R.A.; Furey, W.; Sossenheimer, M.J.; Ulrich, C.D.; Martin, S.P.; Gates, L.K.; Amann, S.T.; Toskes, P.P.; et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 1996, 14, 141–145. [Google Scholar] [CrossRef] [PubMed]
- LaRusch, J.; Whitcomb, D.C. Genetics of pancreatitis. Curr. Opin. Gastroenterol. 2011, 27, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Luck, W.; Hennies, H.C.; Classen, M.; Kage, A.; Lass, U.; Landt, O.; Becker, M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 2000, 25, 213–216. [Google Scholar] [CrossRef]
- Masson, E.; Chen, J.M.; Scotet, V.; Le Maréchal, C.; Férec, C. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum. Genet. 2008, 123, 83–91. [Google Scholar] [CrossRef]
- Rosendahl, J.; Witt, H.; Szmola, R.; Bhatia, E.; Ozsvári, B.; Landt, O.; Schulz, H.U.; Gress, T.M.; Pfützer, R.; Löhr, M.; et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 2008, 40, 78–82. [Google Scholar] [CrossRef]
- Toldi, V.; Szabó, A.; Sahin-Tóth, M. Inactivation of mesotrypsin by chymotrypsin C prevents trypsin inhibitor degradation. J. Biol. Chem. 2020, 295, 3447–3455. [Google Scholar] [CrossRef]
- Sahin-Tóth, M. Genetic risk in chronic pancreatitis: The misfolding-dependent pathway. Curr. Opin. Gastroenterol. 2017, 33, 390–395. [Google Scholar] [CrossRef]
- Witt, H.; Beer, S.; Rosendahl, J.; Chen, J.M.; Chandak, G.R.; Masamune, A.; Bence, M.; Szmola, R.; Oracz, G.; Macek, M.; et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 2013, 45, 1216–1220. [Google Scholar] [CrossRef]
- Kereszturi, E.; Szmola, R.; Kukor, Z.; Simon, P.; Weiss, F.U.; Lerch, M.M.; Sahin-Tóth, M. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: A novel disease mechanism. Hum. Mutat. 2009, 30, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Fjeld, K.; Weiss, F.U.; Lasher, D.; Rosendahl, J.; Chen, J.M.; Johansson, B.B.; Kirsten, H.; Ruffert, C.; Masson, E.; Steine, S.J.; et al. A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat. Genet. 2015, 47, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Sahin-Tóth, M. Channelopathy of the Pancreas Causes Chronic Pancreatitis. Gastroenterology 2020, 158, 1538–1540. [Google Scholar] [CrossRef]
- Hegyi, E.; Sahin-Tóth, M. Genetic Risk in Chronic Pancreatitis: The Trypsin-Dependent Pathway. Dig. Dis. Sci. 2017, 62, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kotani, H.; Sörgel, F.L.; Chen, J.M.; Hamada, S.; Sakaguchi, R.; Masson, E.; Nakano, E.; Kakuta, Y.; Niihori, T.; et al. Variants That Affect Function of Calcium Channel TRPV6 Are Associated With Early-Onset Chronic Pancreatitis. Gastroenterology 2020, 158, 1626–1641.e1628. [Google Scholar] [CrossRef]
- Zou, W.B.; Wang, Y.C.; Ren, X.L.; Wang, L.; Deng, S.J.; Mao, X.T.; Li, Z.S.; Liao, Z. TRPV6 variants confer susceptibility to chronic pancreatitis in the Chinese population. Hum. Mutat. 2020, 41, 1351–1357. [Google Scholar] [CrossRef]
- Howes, N.; Lerch, M.M.; Greenhalf, W.; Stocken, D.D.; Ellis, I.; Simon, P.; Truninger, K.; Ammann, R.; Cavallini, G.; Charnley, R.M.; et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2004, 2, 252–261. [Google Scholar] [CrossRef]
- Keim, V.; Bauer, N.; Teich, N.; Simon, P.; Lerch, M.M.; Mössner, J. Clinical characterization of patients with hereditary pancreatitis and mutations in the cationic trypsinogen gene. Am. J. Med. 2001, 111, 622–626.e1951. [Google Scholar] [CrossRef]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Tóth, M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 2019, 156, 1951–1968.e1951. [Google Scholar] [CrossRef] [PubMed]
- Rebours, V.; Boutron-Ruault, M.C.; Schnee, M.; Férec, C.; Le Maréchal, C.; Hentic, O.; Maire, F.; Hammel, P.; Ruszniewski, P.; Lévy, P. The natural history of hereditary pancreatitis: A national series. Gut 2009, 58, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Joergensen, M.T.; Brusgaard, K.; Crüger, D.G.; Gerdes, A.M.; Schaffalitzky de Muckadell, O.B. Genetic, epidemiological, and clinical aspects of hereditary pancreatitis: A population-based cohort study in Denmark. Am. J. Gastroenterol. 2010, 105, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Applebaum-Shapiro, S.E.; Finch, R.; Pfützer, R.H.; Hepp, L.A.; Gates, L.; Amann, S.; Martin, S.; Ulrich, C.D.; Whitcomb, D.C. Hereditary pancreatitis in North America: The Pittsburgh-Midwest Multi-Center Pancreatic Study Group Study. Pancreatology 2001, 1, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Szabó, A.; Sahin-Tóth, M. Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J. Biol. Chem. 2012, 287, 20701–20710. [Google Scholar] [CrossRef] [PubMed]
- Nemoda, Z.; Sahin-Tóth, M. Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J. Biol. Chem. 2006, 281, 11879–11886. [Google Scholar] [CrossRef]
- Chen, J.M.; Kukor, Z.; Le Maréchal, C.; Tóth, M.; Tsakiris, L.; Raguénès, O.; Férec, C.; Sahin-Tóth, M. Evolution of trypsinogen activation peptides. Mol. Biol. Evol. 2003, 20, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Geisz, A.; Hegyi, P.; Sahin-Tóth, M. Robust autoactivation, chymotrypsin C independence and diminished secretion define a subset of hereditary pancreatitis-associated cationic trypsinogen mutants. FEBS J. 2013, 280, 2888–2899. [Google Scholar] [CrossRef]
- Rygiel, A.M.; Beer, S.; Simon, P.; Wertheim-Tysarowska, K.; Oracz, G.; Kucharzik, T.; Tysarowski, A.; Niepokój, K.; Kierkus, J.; Jurek, M.; et al. Gene conversion between cationic trypsinogen (PRSS1) and the pseudogene trypsinogen 6 (PRSS3P2) in patients with chronic pancreatitis. Hum. Mutat. 2015, 36, 350–356. [Google Scholar] [CrossRef]
- Schnúr, A.; Beer, S.; Witt, H.; Hegyi, P.; Sahin-Tóth, M. Functional effects of 13 rare PRSS1 variants presumed to cause chronic pancreatitis. Gut 2014, 63, 337–343. [Google Scholar] [CrossRef]
- Aoun, E.; Chang, C.C.; Greer, J.B.; Papachristou, G.I.; Barmada, M.M.; Whitcomb, D.C. Pathways to injury in chronic pancreatitis: Decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS ONE 2008, 3, e2003. [Google Scholar] [CrossRef]
- Gasiorowska, A.; Talar-Wojnarowska, R.; Czupryniak, L.; Smolarz, B.; Romanowicz-Makowska, H.; Kulig, A.; Malecka-Panas, E. The prevalence of cationic trypsinogen (PRSS1) and serine protease inhibitor, Kazal type 1 (SPINK1) gene mutations in Polish patients with alcoholic and idiopathic chronic pancreatitis. Dig. Dis. Sci. 2011, 56, 894–901. [Google Scholar] [CrossRef]
- Di Leo, M.; Bianco, M.; Zuppardo, R.A.; Guslandi, M.; Calabrese, F.; Mannucci, A.; Neri, T.M.; Testoni, P.A.; Leandro, G.; Cavestro, G.M. Meta-analysis of the impact of SPINK1 p.N34S gene variation in Caucasic patients with chronic pancreatitis. An update. Dig. Liver Dis. 2017, 49, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Szabó, A.; Toldi, V.; Gazda, L.D.; Demcsák, A.; Tőzsér, J.; Sahin-Tóth, M. Defective binding of SPINK1 variants is an uncommon mechanism for impaired trypsin inhibition in chronic pancreatitis. J. Biol. Chem. 2021, 100343. [Google Scholar] [CrossRef]
- Drenth, J.P.; te Morsche, R.; Jansen, J.B. Mutations in serine protease inhibitor Kazal type 1 are strongly associated with chronic pancreatitis. Gut 2002, 50, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Pfützer, R.H.; Barmada, M.M.; Brunskill, A.P.; Finch, R.; Hart, P.S.; Neoptolemos, J.; Furey, W.F.; Whitcomb, D.C. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 2000, 119, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Apte, M.V.; Keim, V.; Wilson, J.S. Chronic pancreatitis: Challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 2007, 132, 1557–1573. [Google Scholar] [CrossRef]
- Masson, E.; Chen, J.M.; Audrézet, M.P.; Cooper, D.N.; Férec, C. A conservative assessment of the major genetic causes of idiopathic chronic pancreatitis: Data from a comprehensive analysis of PRSS1, SPINK1, CTRC and CFTR genes in 253 young French patients. PLoS ONE 2013, 8, e73522. [Google Scholar] [CrossRef]
- Rygiel, A.M.; Wojnicka-Stolarz, M.; Niepokój, K.; Oracz, G.; Bal, J.; Wertheim-Tysarowska, K.; Gutkowski, K. Chronic Pancreatitis in a Patient With the P.Asn34ser Homozygous Spink1 Mutation--Own Experience. Dev. Period Med. 2015, 19, 347–350. [Google Scholar]
- Kume, K.; Masamune, A.; Kikuta, K.; Shimosegawa, T. [-215G>A.; IVS3+2T>C] mutation in the SPINK1 gene causes exon 3 skipping and loss of the trypsin binding site. Gut 2006, 55, 1214. [Google Scholar] [CrossRef]
- Kereszturi, E.; Király, O.; Sahin-Tóth, M. Minigene analysis of intronic variants in common SPINK1 haplotypes associated with chronic pancreatitis. Gut 2009, 58, 545–549. [Google Scholar] [CrossRef]
- Beer, S.; Zhou, J.; Szabó, A.; Keiles, S.; Chandak, G.R.; Witt, H.; Sahin-Tóth, M. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 2013, 62, 1616–1624. [Google Scholar] [CrossRef]
- Szabó, A.; Ludwig, M.; Hegyi, E.; Szépeová, R.; Witt, H.; Sahin-Tóth, M. Mesotrypsin Signature Mutation in a Chymotrypsin C (CTRC) Variant Associated with Chronic Pancreatitis. J. Biol. Chem. 2015, 290, 17282–17292. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, J.; Landt, O.; Bernadova, J.; Kovacs, P.; Teich, N.; Bödeker, H.; Keim, V.; Ruffert, C.; Mössner, J.; Kage, A.; et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: Is the role of mutated CFTR overestimated? Gut 2013, 62, 582–592. [Google Scholar] [CrossRef]
- Grabarczyk, A.M.; Oracz, G.; Wertheim-Tysarowska, K.; Kujko, A.A.; Wejnarska, K.; Kolodziejczyk, E.; Bal, J.; Koziel, D.; Kowalik, A.; Gluszek, S.; et al. Chymotrypsinogen C Genetic Variants, Including c.180TT, Are Strongly Associated with Chronic Pancreatitis in Pediatric Patients. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 652–657. [Google Scholar] [CrossRef] [PubMed]
- LaRusch, J.; Lozano-Leon, A.; Stello, K.; Moore, A.; Muddana, V.; O'Connell, M.; Diergaarde, B.; Yadav, D.; Whitcomb, D.C. The Common Chymotrypsinogen C (CTRC) Variant G60G (C.180T) Increases Risk of Chronic Pancreatitis But Not Recurrent Acute Pancreatitis in a North American Population. Clin. Transl. Gastroenterol. 2015, 6, e68. [Google Scholar] [CrossRef]
- Kujko, A.A.; Berki, D.M.; Oracz, G.; Wejnarska, K.; Antoniuk, J.; Wertheim-Tysarowska, K.; Kołodziejczyk, E.; Bal, J.; Sahin-Tóth, M.; Rygiel, A.M. A novel p.Ser282Pro. Gut 2017, 66, 1728–1730. [Google Scholar] [CrossRef]
- Németh, B.C.; Patai, Á.; Sahin-Tóth, M.; Hegyi, P. Misfolding cationic trypsinogen variant p.L104P causes hereditary pancreatitis. Gut 2017, 66, 1727–1728. [Google Scholar] [CrossRef]
- Hegyi, E.; Sahin-Tóth, M. Human. Gut 2019, 68, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.B.; Fjeld, K.; El Jellas, K.; Gravdal, A.; Dalva, M.; Tjora, E.; Ræder, H.; Kulkarni, R.N.; Johansson, S.; Njølstad, P.R.; et al. The role of the carboxyl ester lipase (CEL) gene in pancreatic disease. Pancreatology 2018, 18, 12–19. [Google Scholar] [CrossRef]
- Zou, W.B.; Boulling, A.; Masamune, A.; Issarapu, P.; Masson, E.; Wu, H.; Sun, X.T.; Hu, L.H.; Zhou, D.Z.; He, L.; et al. No Association Between CEL-HYB Hybrid Allele and Chronic Pancreatitis in Asian Populations. Gastroenterology 2016, 150, 1558–1560.e1555. [Google Scholar] [CrossRef]
- Dalva, M.; El Jellas, K.; Steine, S.J.; Johansson, B.B.; Ringdal, M.; Torsvik, J.; Immervoll, H.; Hoem, D.; Laemmerhirt, F.; Simon, P.; et al. Copy number variants and VNTR length polymorphisms of the carboxyl-ester lipase (CEL) gene as risk factors in pancreatic cancer. Pancreatology 2017, 17, 83–88. [Google Scholar] [CrossRef]
- Oracz, G.; Kujko, A.A.; Fjeld, K.; Wertheim-Tysarowska, K.; Adamus-Białek, W.; Steine, S.J.; Koziel, D.; Gluszek, S.; Molven, A.; Rygiel, A.M. The hybrid allele 1 of carboxyl-ester lipase (CEL-HYB1) in Polish pediatric patients with chronic pancreatitis. Pancreatology 2019, 19, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, B.M.; Zino, S.; Fjeld, K.; Molven, A.; Lowe, M.E.; Xiao, X. Single nucleotide polymorphisms in CEL-HYB1 increase risk for chronic pancreatitis through proteotoxic misfolding. Hum. Mutat. 2020, 41, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Linsdell, P. Architecture and functional properties of the CFTR channel pore. Cell. Mol. Life Sci. 2017, 74, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Sharer, N.; Schwarz, M.; Malone, G.; Howarth, A.; Painter, J.; Super, M.; Braganza, J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N. Engl. J. Med. 1998, 339, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Cuppens, H.; Macek, M.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef]
- Schneider, A.; Larusch, J.; Sun, X.; Aloe, A.; Lamb, J.; Hawes, R.; Cotton, P.; Brand, R.E.; Anderson, M.A.; Money, M.E.; et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology 2011, 140, 162–171. [Google Scholar] [CrossRef]
- Freeman, A.J.; Ooi, C.Y. Pancreatitis and pancreatic cystosis in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16, S79–S86.e2226. [Google Scholar] [CrossRef] [PubMed]
- Pallagi, P.; Venglovecz, V.; Rakonczay, Z.; Borka, K.; Korompay, A.; Ozsvári, B.; Judák, L.; Sahin-Tóth, M.; Geisz, A.; Schnúr, A.; et al. Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl⁻ channels and luminal anion exchangers. Gastroenterology 2011, 141, 2228–2239.e2226. [Google Scholar] [CrossRef]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef]
- Hegyi, P.; Wilschanski, M.; Muallem, S.; Lukacs, G.L.; Sahin-Tóth, M.; Uc, A.; Gray, M.A.; Rakonczay, Z.; Maléth, J. CFTR: A New Horizon in the Pathomechanism and Treatment of Pancreatitis. Rev. Physiol. Biochem. Pharmacol. 2016, 170, 37–66. [Google Scholar] [CrossRef]
- LaRusch, J.; Jung, J.; General, I.J.; Lewis, M.D.; Park, H.W.; Brand, R.E.; Gelrud, A.; Anderson, M.A.; Banks, P.A.; Conwell, D.; et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet. 2014, 10, e1004376. [Google Scholar] [CrossRef]
- Saotome, K.; Singh, A.K.; Yelshanskaya, M.V.; Sobolevsky, A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature 2016, 534, 506–511. [Google Scholar] [CrossRef]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andréasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Oracz, G.; Zarod, M.; Gambin, T.; Drozak, A.; Kwiatkowski, S.; Wertheim-Tysarowska, K.; Kolodziejczyk, E.; Sawicka, J.; Jackiewicz, M.; Kosińska, J.; et al. TRPV6-defective variants are associated with chronic pancreatitis in Polish pediatric patients. In Proceedings of the 52nd Meeting of the European Pancreatic Club Combined with the International Association of Pancreatology, Paris, France, 1–3 July 2020; pp. S1–S194. [Google Scholar]
- Whitcomb, D.C. Genetic risk factors for pancreatic disorders. Gastroenterology 2013, 144, 1292–1302. [Google Scholar] [CrossRef]
- Noone, P.G.; Zhou, Z.; Silverman, L.M.; Jowell, P.S.; Knowles, M.R.; Cohn, J.A. Cystic fibrosis gene mutations and pancreatitis risk: Relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology 2001, 121, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Férec, C. Chronic pancreatitis: Genetics and pathogenesis. Annu. Rev. Genomics Hum. Genet. 2009, 10, 63–87. [Google Scholar] [CrossRef] [PubMed]
- Rebours, V.; Boutron-Ruault, M.C.; Schnee, M.; Férec, C.; Maire, F.; Hammel, P.; Ruszniewski, P.; Lévy, P. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: A national exhaustive series. Am. J. Gastroenterol. 2008, 103, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Chhoda, A.; Lu, L.; Clerkin, B.M.; Risch, H.; Farrell, J.J. Current Approaches to Pancreatic Cancer Screening. Am. J. Pathol. 2019, 189, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Kadaj-Lipka, R.; Lipiński, M.; Adrych, K.; Durlik, M.; Gąsiorowska, A.; Jarosz, M.; Jurkowska, G.; Małecka-Panas, E.; Oracz, G.; Rosołowski, M.; et al. Diagnostic and therapeutic recommendations for chronic pancreatitis. Recommendations of the Working Group of the Polish Society of Gastroenterology and the Polish Pancreas Club. Prz. Gastroenterol. 2018, 13, 167–181. [Google Scholar] [CrossRef]
- Whitcomb, D.C.; Shimosegawa, T.; Chari, S.T.; Forsmark, C.E.; Frulloni, L.; Garg, P.; Hegyi, P.; Hirooka, Y.; Irisawa, A.; Ishikawa, T.; et al. International consensus statements on early chronic Pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with The International Association of Pancreatology, American Pancreatic Association, Japan Pancreas Society, PancreasFest Working Group and European Pancreatic Club. Pancreatology 2018, 18, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Girodon, E.; Rebours, V.; Chen, J.M.; Pagin, A.; Levy, P.; Ferec, C.; Bienvenu, T. Clinical interpretation of SPINK1 and CTRC variants in pancreatitis. Pancreatology 2020, 20, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Minowa, K.; Nakano, S.; Isayama, H.; Shimizu, T. Genetic Abnormalities in Pancreatitis: An Update on Diagnosis, Clinical Features, and Treatment. Diagnostics 2020, 11, 31. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).