
Ectodermal Dysplasia-Syndactyly Syndrome with Toe-Only Minimal Syndactyly Due to a Novel Mutation in NECTIN4: A Case Report and Literature Review

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
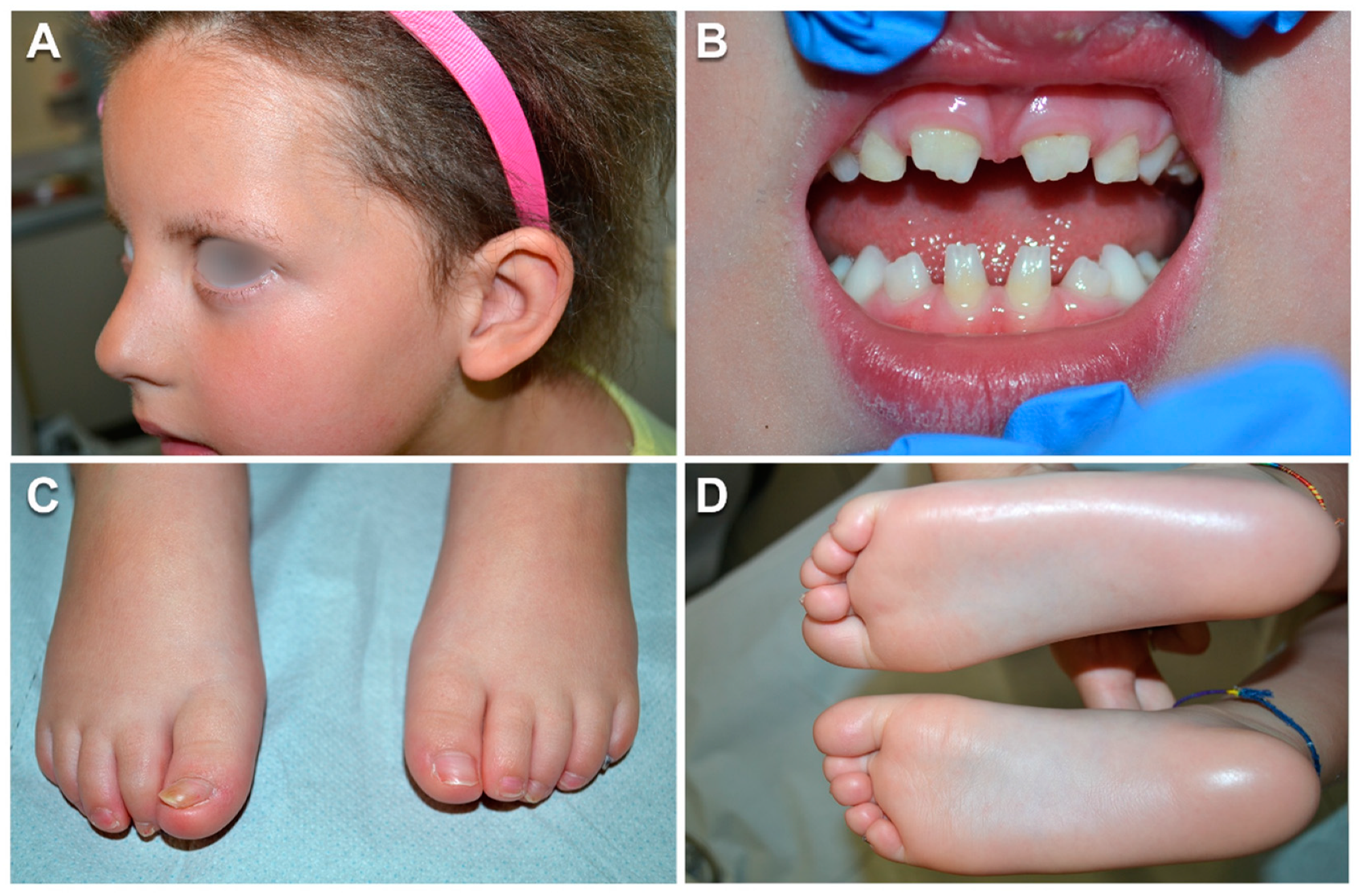
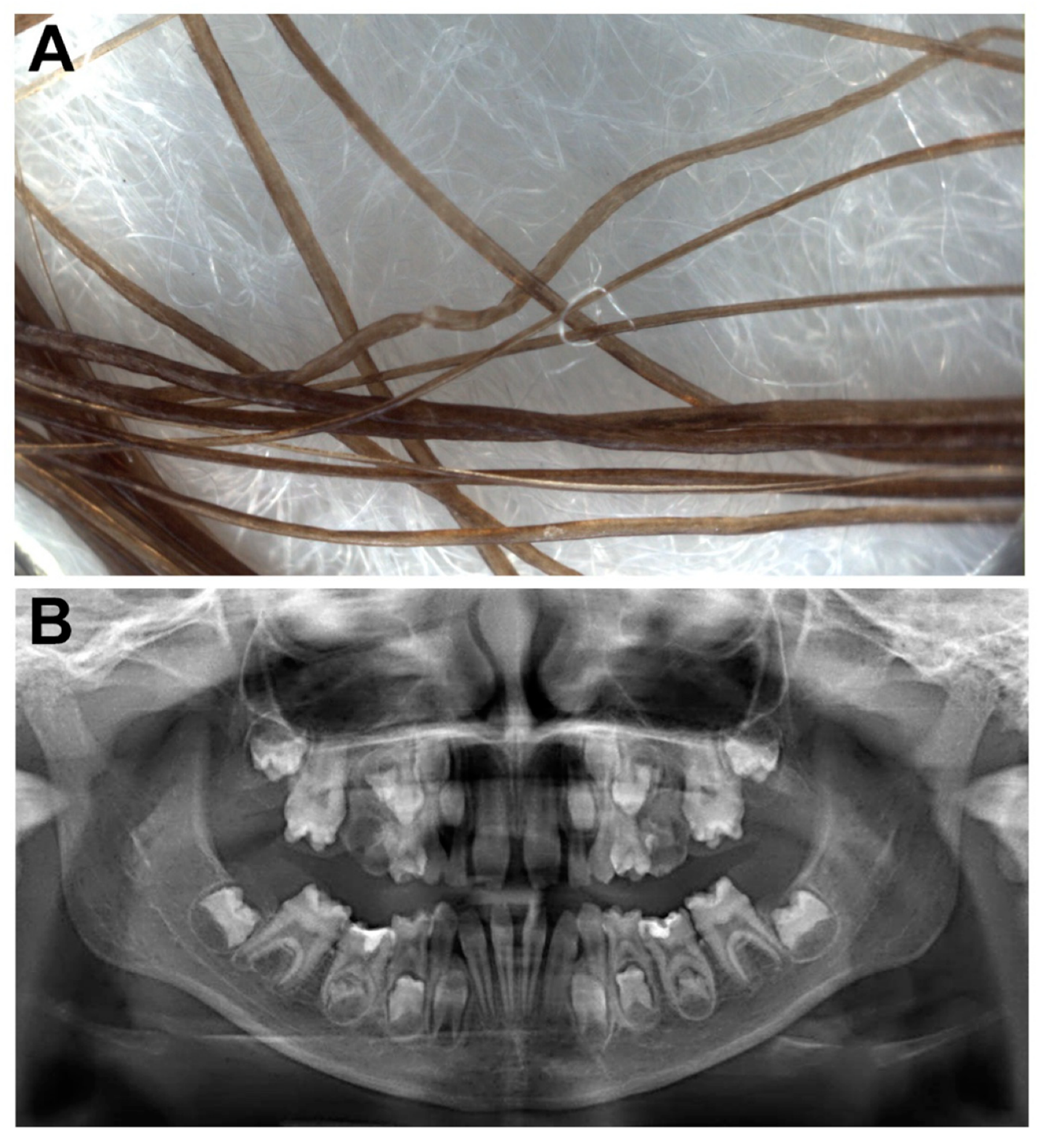
3.1. Case Report
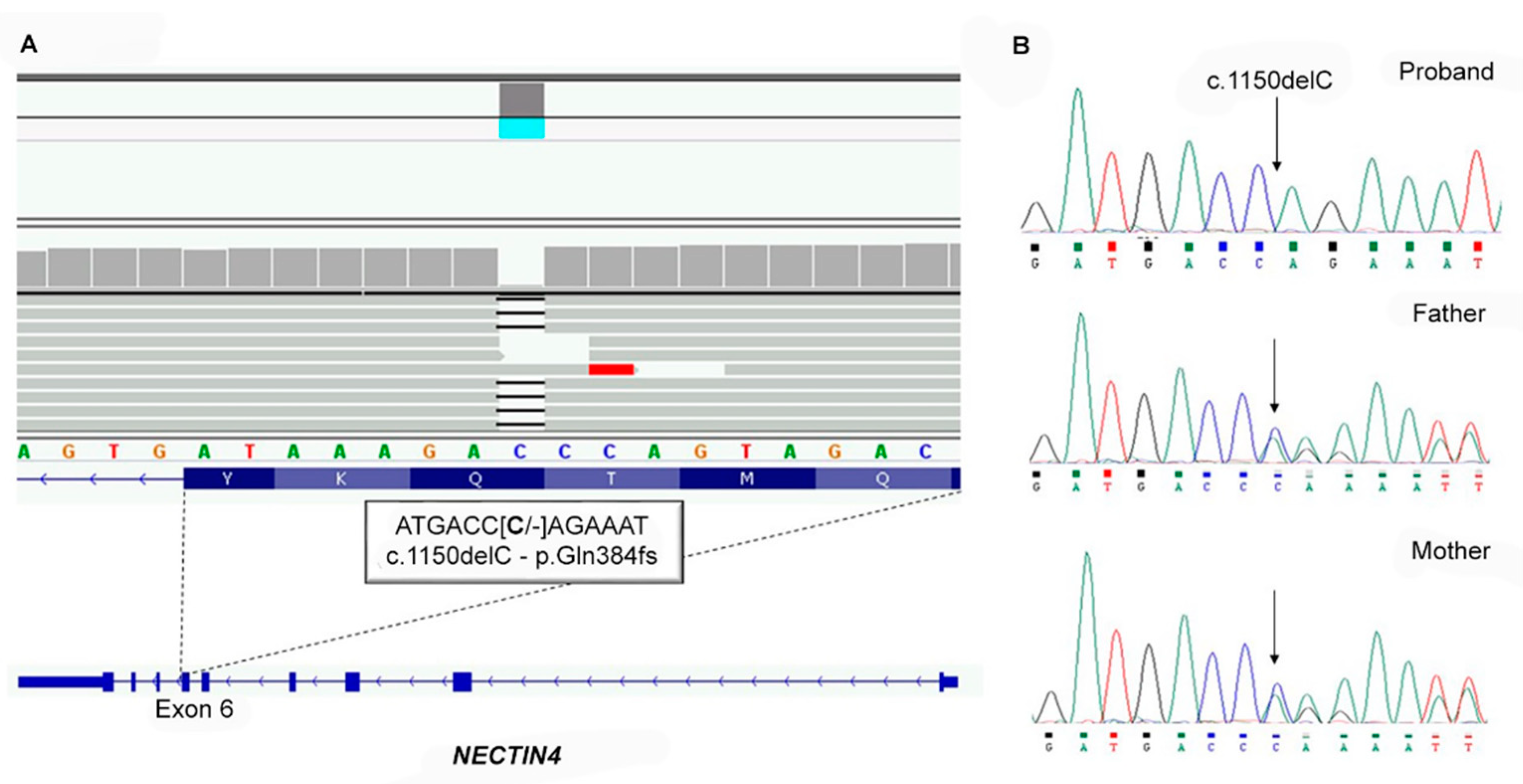
3.2. Molecular Genetic Analysis
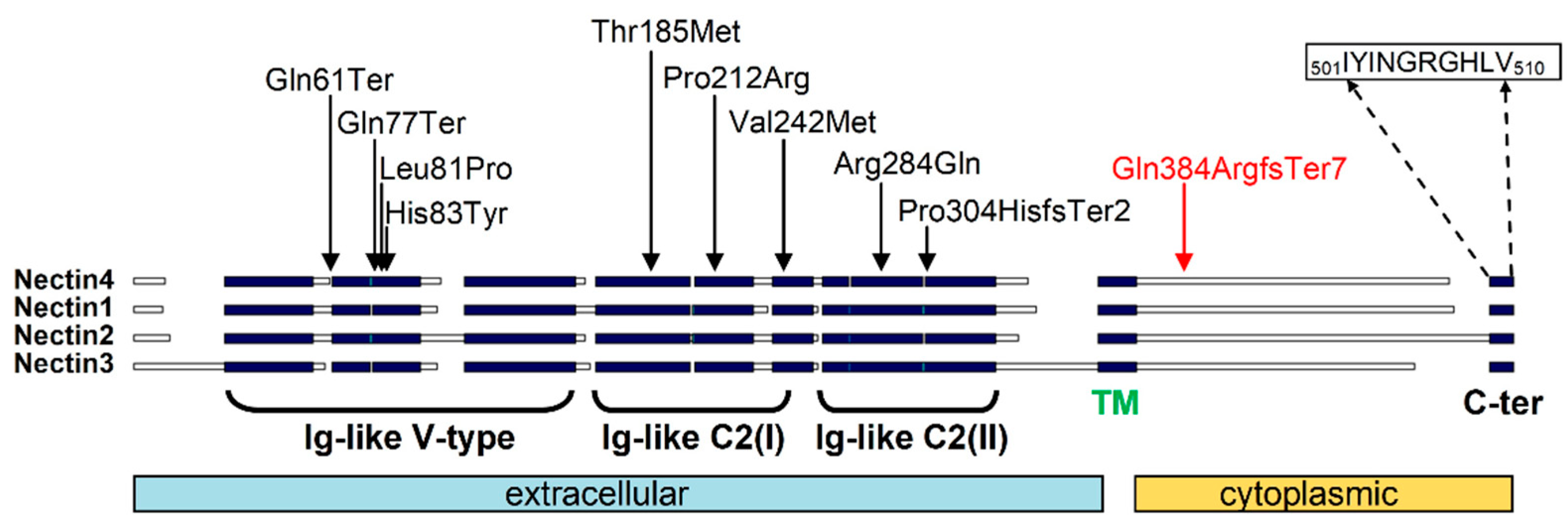
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinheiro, M.; Freire-Maia, N. Ectodermal dysplasias: A clinical classification and a causal review. Am. J. Med. Genet. 1994, 53, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. Part A 2019, 179, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Itin, P.H. Etiology and pathogenesis of ectodermal dysplasias. Am. J. Med. Genet. Part A 2014, 164, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Fortugno, P.; Bottillo, I.; Lopez, M.; Josselin, E.; Boudghene-Stambouli, O.; Agolini, E.; Bernardini, L.; Bellacchio, E.; Iannicelli, M.; et al. Mutations in PVRL4, Encoding Cell Adhesion Molecule Nectin-4, Cause Ectodermal Dysplasia-Syndactyly Syndrome. Am. J. Hum. Genet. 2010, 87, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Agolini, E.; Fortugno, P. Nectinopathies: An emerging group of ectodermal dysplasia syndromes. G. Ital. Dermatol. Venereol. 2013, 148, 59–64. [Google Scholar] [PubMed]
- Florian, R.; Gruber, R.; Volc-Platzer, B. A novel homozygous mutation in PVRL4 causes ectodermal dysplasia-syndactyly syndrome 1. Int. J. Dermatol. 2018, 57, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://euc1.sh.basespace.illumina.com (accessed on 13 May 2021).
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Mandai, K.; Rikitake, Y.; Mori, M.; Takai, Y. Nectins and Nectin-Like Molecules in Development and Disease. Curr. Top. Dev. Biol. 2015, 112, 197–231. [Google Scholar] [CrossRef] [PubMed]
- Samuelov, L.; Sprecher, E.; Paus, R. The role of P-cadherin in skin biology and skin pathology: Lessons from the hair follicle. Cell Tissue Res. 2015, 360, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Hu, D.; Bustos, T.; Zlotogora, J.; Richieri-Costa, A.; Helms, J.A.; Spritz, R.A. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat. Genet. 2000, 25, 427–430. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.; Welsh, N.J.; Braga, V.M.M. Cycling around cell–cell adhesion with Rho GTPase regulators. J. Cell Sci. 2013, 126, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Fortugno, P.; Josselin, E.; Tsiakas, K.; Agolini, E.; Cestra, G.; Teson, M.; Santer, R.; Castiglia, D.; Novelli, G.; Dallapiccola, B.; et al. Nectin-4 Mutations Causing Ectodermal Dysplasia with Syndactyly Perturb the Rac1 Pathway and the Kinetics of Adherens Junction Formation. J. Investig. Dermatol. 2014, 134, 2146–2153. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, D.; Yamada, A.; Amano, T.; Yasuhara, R.; Kimura, A.; Sakahara, M.; Tsumaki, N.; Takeda, S.; Tamura, M.; Nakamura, M.; et al. Essential mesenchymal role of small GTPase Rac1 in interdigital programmed cell death during limb development. Dev. Biol. 2009, 335, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, M.; Henningsen, E.; Hertz, J.; Grejsen, D.; Bygum, A. A Retrospective Study of Clinical and Mutational Findings in 45 Danish Families with Ectodermal Dysplasia. Acta Derm. Venereol. 2014, 94, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Jelani, M.; Chishti, M.S.; Ahmad, W. Mutation in PVRL4 gene encoding nectin-4 underlies ectodermal-dysplasia-syndactyly syndrome (EDSS1). J. Hum. Genet. 2011, 56, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.I.; Dar, R.N.; Shah, A.A.; Ahmad, W. A Novel Homozygous Nonsense Mutation in thePVRL4Gene and Expansion of Clinical Spectrum of EDSS1. Ann. Hum. Genet. 2015, 79, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Dardour, L.; Cosyns, K.; Devriendt, K. A Novel Missense Variant in the PVRL4 Gene Underlying Ectodermal Dysplasia-Syndactyly Syndrome in a Turkish Child. Mol. Syndr. 2017, 9, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Nasir, A.; Thiele, H.; Umair, M.; Borck, G.; Ahmad, W. A novel homozygous missense variant inNECTIN4 (PVRL4)causing ectodermal dysplasia cutaneous syndactyly syndrome. Ann. Hum. Genet. 2018, 82, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; Fabre, S.; Lecocq, E.; Adelaïde, J.; Dubreuil, P.; Lopez, M. Nectin4/PRR4, a New Afadin-associated Member of the Nectin Family That Trans-interacts with Nectin1/PRR1 through V Domain Interaction. J. Biol. Chem. 2001, 276, 43205–43215. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Number | [4] | [16] | [13] | [15] | [17] | [18] | [19] | [6] | Present Case | |
|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | Family 2 | |||||||||
| NECTIN4 variant (cDNA, protein) * | c.851G>A, p.Arge284Gln | c.554C>T, p.Thr185Met; c.906delT p.Pro304HisfsTer2 | c.635C>G, p.Pro212Arg | c.724G>A, p.Val242Met | Exon 2 in-frame deletion | c.181C>T, p.AspGln61Ter | c.247C>T, p.His83Tyr | c.242T>C, p.Leu81Pro | c.229C>T, p.Gln77Ter | c.1150delC, p.Gln384ArgfsTer7 |
| Number of cases | 4 | 2 | 10 | 3 | 1 | 3 | 1 | 4 | 1 | 1 |
| Consanguinity (Y/N) | Y | No | Y | Y | NR | Y | Y | Y | Y | N |
| Origin | Algeria | Italy | Pakistan | Afghanistan | Denmark | Azad Jammu and Kashmir | Turkey | Pakistan | Turkey | Italy |
| Dry skin (Y/N) | NR | NR | NR | NR | N | NR | Y | Y £ | Y | Y £ |
| PPK^ (Y/N) | NR | NR | Y | NR | NR | Y | NR | Y | N | Y |
| Nail dystrophy (Y/N) | N | N | Y | NR | Y | Y | Y | Y | N | Y |
| Hair | ||||||||||
| Hypotrichosis (Y/N) | Y | Y | Y | Y | Y | Y ** | Y | Y | Y | Y |
| Pili torti (Y/N) | Y | Y | N | Y | NR | Y | NR | NR | Y | Y |
| Teeth | ||||||||||
| Enamel defects (Y/N) | N | N | Y | NR | Y | Y | NR | NR | Y | Y |
| Peg/conical (Y/N) | Y | Y | Y | NR | NR | Y | Y | Y | Y | Y |
| Widely spaced (Y/N) | Y | Y | Y | Y | NR | Y | Y | Y | Y | Y |
| Hypodontia (Y/N) | NR | NR | NR | NR | NR | Y | NR | NR | NR | Y |
| Cutaneous syndactyly | ||||||||||
| Fingers (Y/N) | Y | Y | Y | Y | Y ° | Y | Y | Y | N | N |
| Toes (Y/N) | Y | Y | Y | Y | Y ° | Y | Y | Y | Y | Y |
| Heat intolerance | N | N | N | Y § | N | Y | N | NR | N | N |
| Other | N | N | N | N | N | deformed pinnae, purulentconjunctivitis | mildly hypoplastic nipples | large pinnae, pointed nose, thin upper lip | N | ostium secundum atrial septal defect |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotunno, R.; Diociaiuti, A.; Dentici, M.L.; Rinelli, M.; Callea, M.; Retrosi, C.; Zambruno, G.; Bellacchio, E.; El Hachem, M. Ectodermal Dysplasia-Syndactyly Syndrome with Toe-Only Minimal Syndactyly Due to a Novel Mutation in NECTIN4: A Case Report and Literature Review. Genes 2021, 12, 748. https://doi.org/10.3390/genes12050748
Rotunno R, Diociaiuti A, Dentici ML, Rinelli M, Callea M, Retrosi C, Zambruno G, Bellacchio E, El Hachem M. Ectodermal Dysplasia-Syndactyly Syndrome with Toe-Only Minimal Syndactyly Due to a Novel Mutation in NECTIN4: A Case Report and Literature Review. Genes. 2021; 12(5):748. https://doi.org/10.3390/genes12050748
Chicago/Turabian StyleRotunno, Roberta, Andrea Diociaiuti, Maria Lisa Dentici, Martina Rinelli, Michele Callea, Chiara Retrosi, Giovanna Zambruno, Emanuele Bellacchio, and May El Hachem. 2021. "Ectodermal Dysplasia-Syndactyly Syndrome with Toe-Only Minimal Syndactyly Due to a Novel Mutation in NECTIN4: A Case Report and Literature Review" Genes 12, no. 5: 748. https://doi.org/10.3390/genes12050748
APA StyleRotunno, R., Diociaiuti, A., Dentici, M. L., Rinelli, M., Callea, M., Retrosi, C., Zambruno, G., Bellacchio, E., & El Hachem, M. (2021). Ectodermal Dysplasia-Syndactyly Syndrome with Toe-Only Minimal Syndactyly Due to a Novel Mutation in NECTIN4: A Case Report and Literature Review. Genes, 12(5), 748. https://doi.org/10.3390/genes12050748