
Thymoquinone Is a Multitarget Single Epidrug That Inhibits the UHRF1 Protein Complex

 , , ,
, , , 

Abstract
1. Introduction
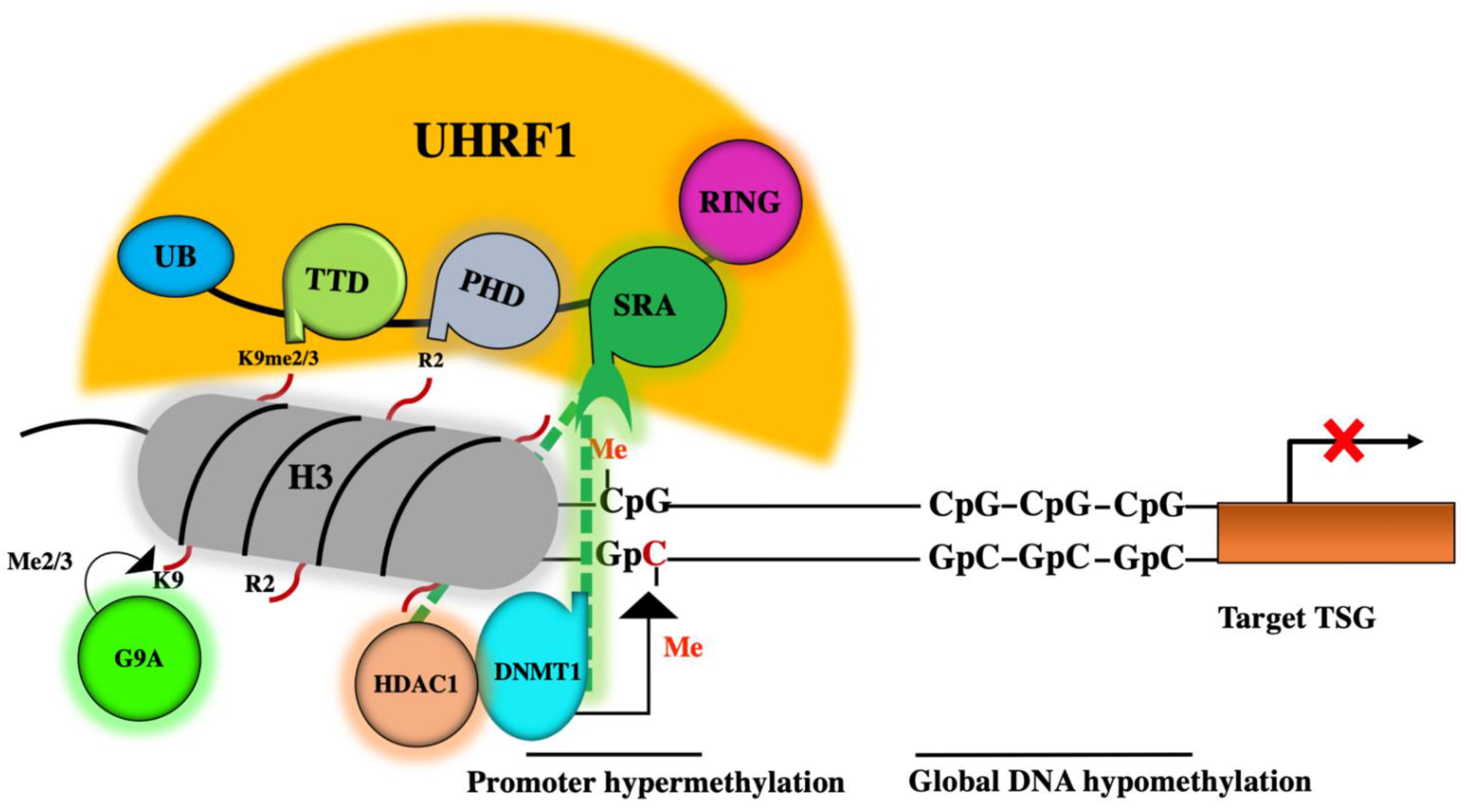
2. Role of the DNMT1/HDAC1/G9a Complex in Epigenetic Silencing of TSGs
3. A master Role for UHRF1 in the ECREM Complex Driving Epigenetic Inhibition of TSGs
4. UHRF1 Is a Main Target of Natural Compounds Exhibiting Anticancer Properties
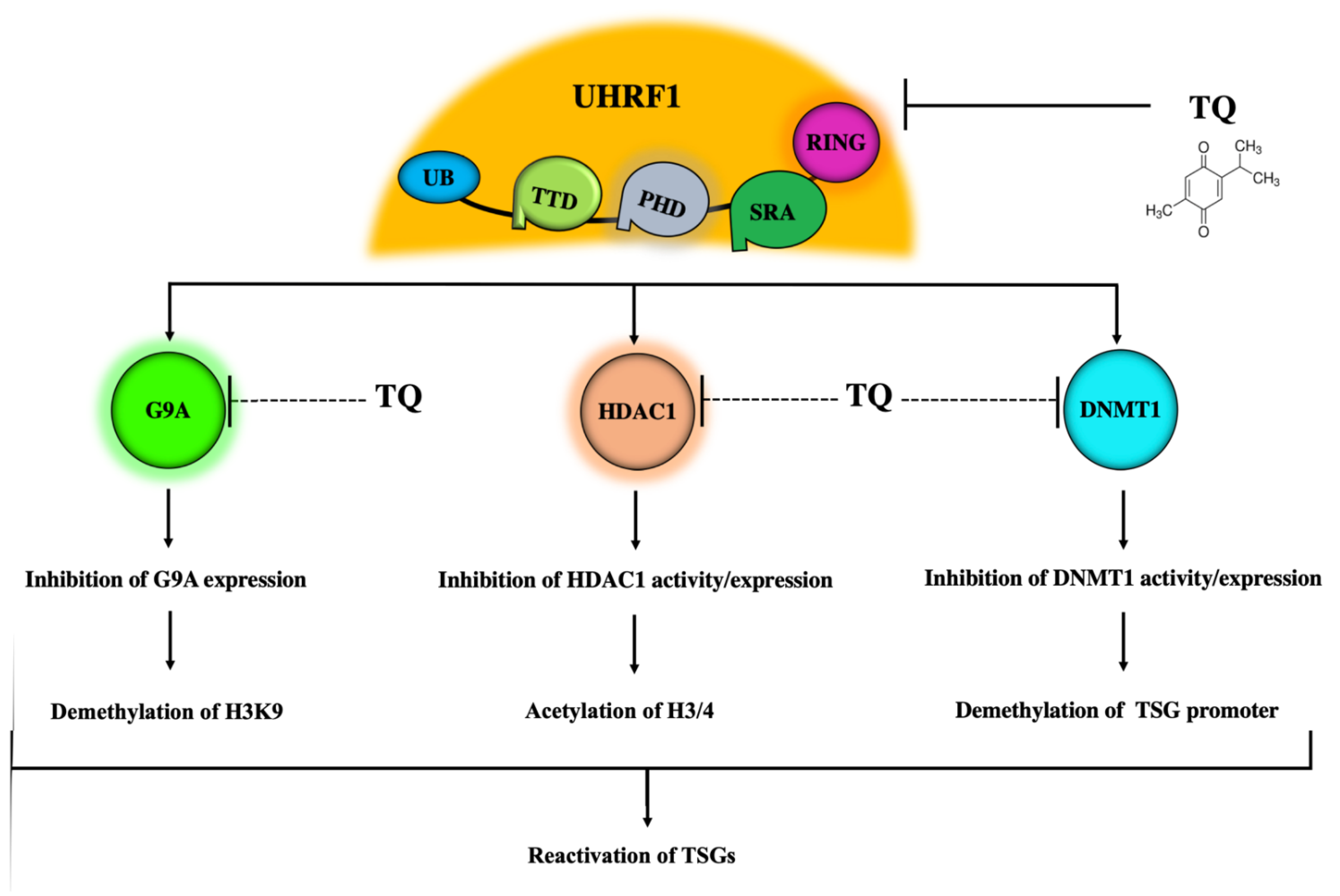
5. The UHRF1 Protein Complex Is a Main Target of TQ
5.1. Inhibitory Effects of TQ on UHRF1
5.2. Inhibitory Effects of TQ on DNMT1 Expression and Activity
5.3. Inhibitory Effects of TQ on HDAC1
5.4. Inhibitory Effects of TQ on G9A
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethical Approval and Consent to Participate
Consent for publication
Abbreviations
| DNMT1 | DNA methyltransferase 1 |
| ECREM | Epigenetic Code Replication Machinery |
| EGCG | Epigallocatechin-3-gallate |
| HAUSP | Herpes virus-Associated Ubiquitin-Specific Protease |
| HDAC1 | Histone deacetylase 1 |
| PDE | Phosphodiesterase |
| PHD | Plant Homeo Domain |
| RING | Really Interesting New Gene domain |
| SRA | Set and Ring Associated domain |
| TSG | Tumor suppressor gene |
| TQ | Thymoquinone |
| TTD | Tandem Tudor Domain |
| UBL | Ubiquitin-like domain |
| UHRF1 | Ubiquitin-like with PHD and RING Finger domains 1 |
References
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epigenetics in breast and prostate cancer. Methods Mol. Biol. 2015, 1238, 425–466. [Google Scholar] [PubMed]
- Lomberk, G.A. Epigenetic silencing of tumor suppressor genes in pancreatic cancer. J. Gastrointest. Cancer 2011, 42, 93–99. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Bronner, C.; Krifa, M.; Mousli, M. Increasing role of UHRF1 in the reading and inheritance of the epigenetic code as well as in tumorogenesis. Biochem. Pharmacol. 2013, 86, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Houliston, R.S.; Lemak, A.; Iqbal, A.; Ivanochko, D.; Duan, S.; Kaustov, L.; Ong, M.S.; Fan, L.; Senisterra, G.; Brown, P.J.; et al. Conformational dynamics of the TTD-PHD histone reader module of the UHRF1 epigenetic regulator reveals multiple histone-binding states, allosteric regulation, and druggability. J. Biol. Chem. 2017, 292, 20947–20959. [Google Scholar] [CrossRef] [PubMed]
- Abhishek, S.; Nivya, M.A.; Nakarakanti, N.K.; Deeksha, W.; Khosla, S.; Rajakumara, E. Biochemical and dynamic basis for combinatorial recognition of H3R2K9me2 by dual domains of UHRF1. Biochimie 2018, 149, 105–114. [Google Scholar] [CrossRef]
- Arita, K.; Isogai, S.; Oda, T.; Unoki, M.; Sugita, K.; Sekiyama, N.; Kuwata, K.; Hamamoto, R.; Tochio, H.; Sato, M.; et al. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc. Natl. Acad. Sci. USA 2012, 109, 12950–12955. [Google Scholar] [CrossRef] [PubMed]
- Kilin, V.; Gavvala, K.; Barthes, N.P.; Michel, B.Y.; Shin, D.; Boudier, C.; Mauffret, O.; Yashchuk, V.; Mousli, M.; Ruff, M.; et al. Dynamics of Methylated Cytosine Flipping by UHRF1. J. Am. Chem. Soc. 2017, 139, 2520–2528. [Google Scholar] [CrossRef]
- Schneider, M.; Trummer, C.; Stengl, A.; Zhang, P.; Szwagierczak, A.; Cardoso, M.C.; Leonhardt, H.; Bauer, C.; Antes, I. Systematic analysis of the binding behaviour of UHRF1 towards different methyl- and carboxylcytosine modification patterns at CpG dyads. PLoS ONE 2020, 15, e0229144. [Google Scholar] [CrossRef]
- Alhosin, M.; Omran, Z.; Zamzami, M.A.; Al-Malki, A.L.; Choudhry, H.; Mousli, M.; Bronner, C. Signalling pathways in UHRF1-dependent regulation of tumor suppressor genes in cancer. J. Exp. Clin. Cancer Res. CR 2016, 35, 174. [Google Scholar] [CrossRef] [PubMed]
- Alhosin, M.; Sharif, T.; Mousli, M.; Etienne-Selloum, N.; Fuhrmann, G.; Schini-Kerth, V.B.; Bronner, C. Down-regulation of UHRF1, associated with re-expression of tumor suppressor genes, is a common feature of natural compounds exhibiting anti-cancer properties. J. Exp. Clin. Cancer Res. CR 2011, 30, 41. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, W.; Ibrahim, A.; Alhosin, M.; Zaayter, L.; Ouararhni, K.; Papin, C.; Ahmad, T.; Hamiche, A.; Mély, Y.; Bronner, C.; et al. The epigenetic integrator UHRF1: On the road to become a universal biomarker for cancer. Oncotarget 2017, 8, 51946–51962. [Google Scholar] [CrossRef]
- Sidhu, H.; Capalash, N. UHRF1: The key regulator of epigenetics and molecular target for cancer therapeutics. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317692205. [Google Scholar] [CrossRef]
- Achour, M.; Jacq, X.; Rondé, P.; Alhosin, M.; Charlot, C.; Chataigneau, T.; Jeanblanc, M.; Macaluso, M.; Giordano, A.; Hughes, A.D.; et al. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene 2008, 27, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Horton, J.R.; Zhang, X.; Bostick, M.; Jacobsen, S.E.; Cheng, X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature 2008, 455, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Unoki, M.; Nishidate, T.; Nakamura, Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene 2004, 23, 7601–7610. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, W.; Bronner, C.; Zaayter, L.; Ahmad, T.; Richert, L.; Alhosin, M.; Ibrahim, A.; Hamiche, A.; Mely, Y.; Mousli, M. Interaction of the epigenetic integrator UHRF1 with the MYST domain of TIP60 inside the cell. J. Exp. Clin. Cancer Res. CR 2017, 36, 188. [Google Scholar] [CrossRef]
- Achour, M.; Fuhrmann, G.; Alhosin, M.; Rondé, P.; Chataigneau, T.; Mousli, M.; Schini-Kerth, V.B.; Bronner, C. UHRF1 recruits the histone acetyltransferase Tip60 and controls its expression and activity. Biochem. Biophys. Res. Commun. 2009, 390, 523–528. [Google Scholar] [CrossRef]
- Kim, J.K.; Estève, P.O.; Jacobsen, S.E.; Pradhan, S. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 2009, 37, 493–505. [Google Scholar] [CrossRef]
- Bronner, C.; Alhosin, M.; Hamiche, A.; Mousli, M. Coordinated Dialogue between UHRF1 and DNMT1 to Ensure Faithful Inheritance of Methylated DNA Patterns. Genes 2019, 10, 65. [Google Scholar] [CrossRef]
- Ibrahim, A.; Alhosin, M.; Papin, C.; Ouararhni, K.; Omran, Z.; Zamzami, M.A.; Al-Malki, A.L.; Choudhry, H.; Mély, Y.; Hamiche, A.; et al. Thymoquinone challenges UHRF1 to commit auto-ubiquitination: A key event for apoptosis induction in cancer cells. Oncotarget 2018, 9, 28599–28611. [Google Scholar] [CrossRef] [PubMed]
- Achour, M.; Mousli, M.; Alhosin, M.; Ibrahim, A.; Peluso, J.; Muller, C.D.; Schini-Kerth, V.B.; Hamiche, A.; Dhe-Paganon, S.; Bronner, C. Epigallocatechin-3-gallate up-regulates tumor suppressor gene expression via a reactive oxygen species-dependent down-regulation of UHRF1. Biochem. Biophys. Res. Commun. 2013, 430, 208–212. [Google Scholar] [CrossRef]
- Liu, X.; Ou, H.; Xiang, L.; Li, X.; Huang, Y.; Yang, D. Elevated UHRF1 expression contributes to poor prognosis by promoting cell proliferation and metastasis in hepatocellular carcinoma. Oncotarget 2017, 8, 10510–10522. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.C.; Lin, G.H.; Wang, B.; Yan, M.; He, B.; Zhang, W.; Yang, A.K.; Long, Z.J.; Liu, Q. UHRF1 suppression promotes cell differentiation and reduces inflammatory reaction in anaplastic thyroid cancer. Oncotarget 2018, 9, 31945–31957. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, J.; Yu, L.; Zhang, Z.; Xu, F.; Jiang, L.; Zhou, X.; He, S. Regulation of RIP3 by the transcription factor Sp1 and the epigenetic regulator UHRF1 modulates cancer cell necroptosis. Cell Death Dis. 2017, 8, e3084. [Google Scholar] [CrossRef]
- Ge, T.T.; Yang, M.; Chen, Z.; Lou, G.; Gu, T. UHRF1 gene silencing inhibits cell proliferation and promotes cell apoptosis in human cervical squamous cell carcinoma CaSki cells. J. Ovarian Res. 2016, 9, 42. [Google Scholar] [CrossRef]
- Matsushita, R.; Yoshino, H.; Enokida, H.; Goto, Y.; Miyamoto, K.; Yonemori, M.; Inoguchi, S.; Nakagawa, M.; Seki, N. Regulation of UHRF1 by dual-strand tumor-suppressor microRNA-145 (miR-145-5p and miR-145-3p): Inhibition of bladder cancer cell aggressiveness. Oncotarget 2016, 7, 28460–28487. [Google Scholar] [CrossRef]
- Wan, X.; Yang, S.; Huang, W.; Wu, D.; Chen, H.; Wu, M.; Li, J.; Li, T.; Li, Y. UHRF1 overexpression is involved in cell proliferation and biochemical recurrence in prostate cancer after radical prostatectomy. J. Exp. Clin. Cancer Res. CR 2016, 35, 34. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, J.; Gong, W.; Zhang, M.; Tang, Z.; Zhang, J.; Quan, Z. UHRF1 depletion suppresses growth of gallbladder cancer cells through induction of apoptosis and cell cycle arrest. Oncol. Rep. 2014, 31, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Niinuma, T.; Kitajima, H.; Kai, M.; Yamamoto, E.; Yorozu, A.; Ishiguro, K.; Sasaki, H.; Sudo, G.; Toyota, M.; Hatahira, T.; et al. UHRF1 depletion and HDAC inhibition reactivate epigenetically silenced genes in colorectal cancer cells. Clin. Epigenet. 2019, 11, 70. [Google Scholar] [CrossRef]
- Seo, J.S.; Choi, Y.H.; Moon, J.W.; Kim, H.S.; Park, S.H. Hinokitiol induces DNA demethylation via DNMT1 and UHRF1 inhibition in colon cancer cells. BMC Cell Biol. 2017, 18, 14. [Google Scholar] [CrossRef]
- Gao, S.P.; Sun, H.F.; Li, L.D.; Fu, W.Y.; Jin, W. UHRF1 promotes breast cancer progression by suppressing KLF17 expression by hypermethylating its promoter. Am. J. Cancer Res. 2017, 7, 1554–1565. [Google Scholar]
- Zhang, Q.; Qiao, L.; Wang, X.; Ding, C.; Chen, J.J. UHRF1 epigenetically down-regulates UbcH8 to inhibit apoptosis in cervical cancer cells. Cell Cycle 2018, 17, 300–308. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Estève, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef]
- Foster, B.M.; Stolz, P.; Mulholland, C.B.; Montoya, A.; Kramer, H.; Bultmann, S.; Bartke, T. Critical Role of the UBL Domain in Stimulating the E3 Ubiquitin Ligase Activity of UHRF1 toward Chromatin. Mol. Cell 2018, 72, 739–752.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, L.; Du, Y.; Xie, S.; Yang, X.; Lian, F.; Zhou, Z.; Qian, C. Structural and mechanistic insights into UHRF1-mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids Res. 2018, 46, 3218–3231. [Google Scholar] [CrossRef] [PubMed]
- Felle, M.; Joppien, S.; Németh, A.; Diermeier, S.; Thalhammer, V.; Dobner, T.; Kremmer, E.; Kappler, R.; Längst, G. The USP7/Dnmt1 complex stimulates the DNA methylation activity of Dnmt1 and regulates the stability of UHRF1. Nucleic Acids Res. 2011, 39, 8355–8365. [Google Scholar] [CrossRef]
- Du, S.; Xu, G.; Zou, W.; Xiang, T.; Luo, Z. Effect of dihydroartemisinin on UHRF1 gene expression in human prostate cancer PC-3 cells. Anti-Cancer Drugs 2017, 28, 384–391. [Google Scholar] [CrossRef]
- Babbio, F.; Pistore, C.; Curti, L.; Castiglioni, I.; Kunderfranco, P.; Brino, L.; Oudet, P.; Seiler, R.; Thalman, G.N.; Roggero, E.; et al. The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene 2012, 31, 4878–4887. [Google Scholar] [CrossRef]
- Estève, P.O.; Chin, H.G.; Smallwood, A.; Feehery, G.R.; Gangisetty, O.; Karpf, A.R.; Carey, M.F.; Pradhan, S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006, 20, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Darakhshan, S.; Bidmeshki Pour, A.; Hosseinzadeh Colagar, A.; Sisakhtnezhad, S. Thymoquinone and its therapeutic potentials. Pharmacol. Res. 2015, 95–96, 138–158. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Tania, M.; Fu, J. Epigenetic role of thymoquinone: Impact on cellular mechanism and cancer therapeutics. Drug Discov. Today 2019, 24, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, Y.K.; Abdelrazek, H.M.A. Cancer: Thymoquinone antioxidant/pro-oxidant effect as potential anticancer remedy. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 115, 108783. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Ahn, K.S.; Hsu, A.; Woo, C.C.; Yuan, Y.; Tan, K.H.B.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; Koh, A.P.F.; et al. Thymoquinone Inhibits Bone Metastasis of Breast Cancer Cells Through Abrogation of the CXCR4 Signaling Axis. Front. Pharmacol. 2018, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Kim, G.H.; Park, E.J.; Oh, T.I.; Lee, S.; Kan, S.Y.; Kang, H.; Kim, B.M.; Kim, J.H.; Lim, J.H. Thymoquinone Selectively Kills Hypoxic Renal Cancer Cells by Suppressing HIF-1α-Mediated Glycolysis. Int. J. Mol. Sci. 2019, 20, 1092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, Y.; Huang, S.; Wang, G.; Han, R.; Lei, F.; Luo, A.; Jing, X.; Zhao, L.; Gu, S.; et al. Thymoquinone inhibits the metastasis of renal cell cancer cells by inducing autophagy via AMPK/mTOR signaling pathway. Cancer Sci. 2018, 109, 3865–3873. [Google Scholar] [CrossRef] [PubMed]
- Ndreshkjana, B.; Çapci, A.; Klein, V.; Chanvorachote, P.; Muenzner, J.K.; Huebner, K.; Steinmann, S.; Erlenbach-Wuensch, K.; Geppert, C.I.; Agaimy, A.; et al. Combination of 5-fluorouracil and thymoquinone targets stem cell gene signature in colorectal cancer cells. Cell Death Dis. 2019, 10, 379. [Google Scholar] [CrossRef]
- Goyal, S.N.; Prajapati, C.P.; Gore, P.R.; Patil, C.R.; Mahajan, U.B.; Sharma, C.; Talla, S.P.; Ojha, S.K. Therapeutic Potential and Pharmaceutical Development of Thymoquinone: A Multitargeted Molecule of Natural Origin. Front. Pharmacol. 2017, 8, 656. [Google Scholar] [CrossRef]
- Woo, C.C.; Kumar, A.P.; Sethi, G.; Tan, K.H. Thymoquinone: Potential cure for inflammatory disorders and cancer. Biochem. Pharmacol. 2012, 83, 443–451. [Google Scholar] [CrossRef]
- Alhosin, M.; Ibrahim, A.; Boukhari, A.; Sharif, T.; Gies, J.P.; Auger, C.; Schini-Kerth, V.B. Anti-neoplastic agent thymoquinone induces degradation of α and β tubulin proteins in human cancer cells without affecting their level in normal human fibroblasts. Investig. New Drugs 2012, 30, 1813–1819. [Google Scholar] [CrossRef]
- Feng, L.M.; Wang, X.F.; Huang, Q.X. Thymoquinone induces cytotoxicity and reprogramming of EMT in gastric cancer cells by targeting PI3K/Akt/mTOR pathway. J. Biosci. 2017, 42, 547–554. [Google Scholar] [CrossRef]
- Paramasivam, A.; Raghunandhakumar, S.; Priyadharsini, J.V.; Jayaraman, G. In Vitro Anti-Neuroblastoma Activity of Thymoquinone against Neuro-2a Cells via Cell-cycle Arrest. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 8313–8319. [Google Scholar] [CrossRef] [PubMed]
- Racoma, I.O.; Meisen, W.H.; Wang, Q.E.; Kaur, B.; Wani, A.A. Thymoquinone inhibits autophagy and induces cathepsin-mediated, caspase-independent cell death in glioblastoma cells. PLoS ONE 2013, 8, e72882. [Google Scholar] [CrossRef] [PubMed]
- Alaufi, O.M.; Noorwali, A.; Zahran, F.; Al-Abd, A.M.; Al-Attas, S. Cytotoxicity of thymoquinone alone or in combination with cisplatin (CDDP) against oral squamous cell carcinoma in vitro. Sci. Rep. 2017, 7, 13131. [Google Scholar] [CrossRef] [PubMed]
- Abukhader, M.M. Thymoquinone in the clinical treatment of cancer: Fact or fiction? Pharmacogn. Rev. 2013, 7, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Odeh, F.; Ismail, S.I.; Abu-Dahab, R.; Mahmoud, I.S.; Al Bawab, A. Thymoquinone in liposomes: A study of loading efficiency and biological activity towards breast cancer. Drug Deliv. 2012, 19, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Ganea, G.M.; Fakayode, S.O.; Losso, J.N.; van Nostrum, C.F.; Sabliov, C.M.; Warner, I.M. Delivery of phytochemical thymoquinone using molecular micelle modified poly(D, L lactide-co-glycolide) (PLGA) nanoparticles. Nanotechnology 2010, 21, 285104. [Google Scholar] [CrossRef]
- Qadi, S.A.; Hassan, M.A.; Sheikh, R.A.; Baothman, O.A.; Zamzami, M.A.; Choudhry, H.; Al-Malki, A.L.; Albukhari, A.; Alhosin, M. Thymoquinone-Induced Reactivation of Tumor Suppressor Genes in Cancer Cells Involves Epigenetic Mechanisms. Epigenetics Insights 2019, 12, 2516865719839011. [Google Scholar] [CrossRef]
- Alhosin, M.; Razvi, S.S.I.; Sheikh, R.A.; Khan, J.A.; Zamzami, M.A.; Choudhry, H. Thymoquinone and Difluoromethylornithine (DFMO) Synergistically Induce Apoptosis of Human Acute T Lymphoblastic Leukemia Jurkat Cells Through the Modulation of Epigenetic Pathways. Technol. Cancer Res. Treat. 2020, 19, 1533033820947489. [Google Scholar] [CrossRef]
- Abusnina, A.; Alhosin, M.; Keravis, T.; Muller, C.D.; Fuhrmann, G.; Bronner, C.; Lugnier, C. Down-regulation of cyclic nucleotide phosphodiesterase PDE1A is the key event of p73 and UHRF1 deregulation in thymoquinone-induced acute lymphoblastic leukemia cell apoptosis. Cell. Signal. 2011, 23, 152–160. [Google Scholar] [CrossRef]
- Alhosin, M.; Abusnina, A.; Achour, M.; Sharif, T.; Muller, C.; Peluso, J.; Chataigneau, T.; Lugnier, C.; Schini-Kerth, V.B.; Bronner, C.; et al. Induction of apoptosis by thymoquinone in lymphoblastic leukemia Jurkat cells is mediated by a p73-dependent pathway which targets the epigenetic integrator UHRF1. Biochem. Pharmacol. 2010, 79, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Shen, N.; Yan, F.; Zhao, N.; Dou, L.; Wu, L.C.; Seiler, C.L.; Yu, L.; Yang, K.; Bachanova, V.; et al. Thymoquinone exerts potent growth-suppressive activity on leukemia through DNA hypermethylation reversal in leukemia cells. Oncotarget 2017, 8, 34453–34467. [Google Scholar] [CrossRef] [PubMed]
- Relles, D.; Chipitsyna, G.I.; Gong, Q.; Yeo, C.J.; Arafat, H.A. Thymoquinone Promotes Pancreatic Cancer Cell Death and Reduction of Tumor Size through Combined Inhibition of Histone Deacetylation and Induction of Histone Acetylation. Adv. Prev. Med. 2016, 2016, 1407840. [Google Scholar] [CrossRef] [PubMed]
- Parbin, S.; Shilpi, A.; Kar, S.; Pradhan, N.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Insights into the molecular interactions of thymoquinone with histone deacetylase: Evaluation of the therapeutic intervention potential against breast cancer. Mol. Biosyst. 2016, 12, 48–58. [Google Scholar] [CrossRef]
- Benedetti, R.; Conte, M.; Iside, C.; Altucci, L. Epigenetic-based therapy: From single- to multi-target approaches. Int. J. Biochem. Cell Biol. 2015, 69, 121–131. [Google Scholar] [CrossRef]
- Ganesan, A. Multitarget Drugs: An Epigenetic Epiphany. ChemMedChem 2016, 11, 1227–1241. [Google Scholar] [CrossRef]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Cedar, H. Epigenetic crosstalk. Mol. Cell 2001, 8, 933–935. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Vaissière, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Bronner, C.; Fuhrmann, G.; Chédin, F.L.; Macaluso, M.; Dhe-Paganon, S. UHRF1 Links the Histone code and DNA Methylation to ensure Faithful Epigenetic Memory Inheritance. Genet. Epigenetics 2010, 2009, 29–36. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef]
- Chen, Y.F.; Luo, R.Z.; Li, Y.; Cui, B.K.; Song, M.; Yang, A.K.; Chen, W.K. High expression levels of COX-2 and P300 are associated with unfavorable survival in laryngeal squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 2013, 270, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Horikoshi, M. Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells Devoted Mol. Cell. Mech. 1998, 3, 789–800. [Google Scholar] [CrossRef]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Judes, G.; Rifaï, K.; Ngollo, M.; Daures, M.; Bignon, Y.J.; Penault-Llorca, F.; Bernard-Gallon, D. A bivalent role of TIP60 histone acetyl transferase in human cancer. Epigenomics 2015, 7, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Stacy, A.J.; Zhang, J.; Craig, M.P.; Hira, A.; Dole, N.; Kadakia, M.P. TIP60 up-regulates ΔNp63α to promote cellular proliferation. J. Biol. Chem. 2019, 294, 17007–17016. [Google Scholar] [CrossRef]
- Shi, X.; Fan, M. Tip60-dependent acetylation of KDM2B promotes osteosarcoma carcinogenesis. J. Cell. Mol. Med. 2019, 23, 6154–6163. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, J.; Chen, T.; Tao, Z.; Zhang, X.; Tian, F.; Zhou, X.; Lu, D. Tat-interactive Protein-60KDA (TIP60) Regulates the Tumorigenesis of Lung Cancer In Vitro. J. Cancer 2017, 8, 2277–2281. [Google Scholar] [CrossRef][Green Version]
- Judes, G.; Dubois, L.; Rifaï, K.; Idrissou, M.; Mishellany, F.; Pajon, A.; Besse, S.; Daures, M.; Degoul, F.; Bignon, Y.J.; et al. TIP60: An actor in acetylation of H3K4 and tumor development in breast cancer. Epigenomics 2018, 10, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.L.; Song, X.; Pei, L.; Liu, L.; Wang, H.; Jia, M. Histone deacetylase HDAC1 expression correlates with the progression and prognosis of lung cancer: A meta-analysis. Medicine 2017, 96, e7663. [Google Scholar] [CrossRef]
- Phi van, D.K.; Mühlbauer, E.; Phi-van, L. Histone deacetylase HDAC1 downregulates transcription of the serotonin transporter (5-HTT) gene in tumor cells. Biochim. Biophys. Acta 2015, 1849, 909–918. [Google Scholar] [CrossRef]
- Liu, X.; Yu, Y.; Zhang, J.; Lu, C.; Wang, L.; Liu, P.; Song, H. HDAC1 Silencing in Ovarian Cancer Enhances the Chemotherapy Response. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 1505–1518. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef]
- Fritzsche, F.R.; Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Scholman, K.; Denkert, C.; Dietel, M.; et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 2008, 8, 381. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef]
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C.; et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol. Cell. Biol. 2007, 27, 4784–4795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bu, L.; Hu, J.; Xu, Z.; Ruan, L.; Fang, Y.; Wang, P. HDAC1 knockdown inhibits invasion and induces apoptosis in non-small cell lung cancer cells. Biol. Chem. 2018, 399, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Krusche, C.A.; Wülfing, P.; Kersting, C.; Vloet, A.; Böcker, W.; Kiesel, L.; Beier, H.M.; Alfer, J. Histone deacetylase-1 and -3 protein expression in human breast cancer: A tissue microarray analysis. Breast Cancer Res. Treat. 2005, 90, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Wisnieski, F.; Calcagno, D.Q.; Leal, M.F.; Chen, E.S.; Gigek, C.O.; Santos, L.C.; Pontes, T.B.; Rasmussen, L.T.; Payão, S.L.; Assumpção, P.P.; et al. Differential expression of histone deacetylase and acetyltransferase genes in gastric cancer and their modulation by trichostatin A. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 6373–6381. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 lysine 4 methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Cao, H.; Li, L.; Yang, D.; Zeng, L.; Yewei, X.; Yu, B.; Liao, G.; Chen, J. Recent progress in histone methyltransferase (G9a) inhibitors as anticancer agents. Eur. J. Med. Chem. 2019, 179, 537–546. [Google Scholar] [CrossRef]
- Yin, C.; Ke, X.; Zhang, R.; Hou, J.; Dong, Z.; Wang, F.; Zhang, K.; Zhong, X.; Yang, L.; Cui, H. G9a promotes cell proliferation and suppresses autophagy in gastric cancer by directly activating mTOR. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 14036–14050. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. TIG 2016, 32, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Scheer, S.; Zaph, C. The Lysine Methyltransferase G9a in Immune Cell Differentiation and Function. Front. Immunol. 2017, 8, 429. [Google Scholar] [CrossRef]
- Mayr, C.; Helm, K.; Jakab, M.; Ritter, M.; Shrestha, R.; Makaju, R.; Wagner, A.; Pichler, M.; Beyreis, M.; Staettner, S.; et al. The histone methyltransferase G9a: A new therapeutic target in biliary tract cancer. Hum. Pathol. 2018, 72, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chiu, D.K.; Tsang, F.H.; Law, C.T.; Cheng, C.L.; Au, S.L.; Lee, J.M.; Wong, C.C.; Ng, I.O.; Wong, C.M. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef]
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907. [Google Scholar] [CrossRef]
- Wozniak, R.J.; Klimecki, W.T.; Lau, S.S.; Feinstein, Y.; Futscher, B.W. 5-Aza-2’-deoxycytidine-mediated reductions in G9A histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene 2007, 26, 77–90. [Google Scholar] [CrossRef]
- Hua, K.T.; Wang, M.Y.; Chen, M.W.; Wei, L.H.; Chen, C.K.; Ko, C.H.; Jeng, Y.M.; Sung, P.L.; Jan, Y.H.; Hsiao, M.; et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef]
- Duvall-Noelle, N.; Karwandyar, A.; Richmond, A.; Raman, D. LASP-1: A nuclear hub for the UHRF1-DNMT1-G9a-Snail1 complex. Oncogene 2016, 35, 1122–1133. [Google Scholar] [CrossRef]
- Bárcena-Varela, M.; Caruso, S.; Llerena, S.; Álvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Rebouissou, S.; Alvarez, L.; Jimenez, M.; et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology 2019, 69, 587–603. [Google Scholar] [CrossRef]
- Cartron, P.F.; Blanquart, C.; Hervouet, E.; Gregoire, M.; Vallette, F.M. HDAC1-mSin3a-NCOR1, Dnmt3b-HDAC1-Egr1 and Dnmt1-PCNA-UHRF1-G9a regulate the NY-ESO1 gene expression. Mol. Oncol. 2013, 7, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Borutinskaitė, V.; Virkšaitė, A.; Gudelytė, G.; Navakauskienė, R. Green tea polyphenol EGCG causes anti-cancerous epigenetic modulations in acute promyelocytic leukemia cells. Leuk. Lymphoma 2018, 59, 469–478. [Google Scholar] [CrossRef]
- Macaluso, M.; Cinti, C.; Russo, G.; Russo, A.; Giordano, A. pRb2/p130-E2F4/5-HDAC1-SUV39H1-p300 and pRb2/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-alpha in breast cancer. Oncogene 2003, 22, 3511–3517. [Google Scholar] [CrossRef]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Li, Y.; Duan, S.; Bronner, C.; Arrowsmith, C.H.; Dhe-Paganon, S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 2008, 455, 822–825. [Google Scholar] [CrossRef]
- Misaki, T.; Yamaguchi, L.; Sun, J.; Orii, M.; Nishiyama, A.; Nakanishi, M. The replication foci targeting sequence (RFTS) of DNMT1 functions as a potent histone H3 binding domain regulated by autoinhibition. Biochem. Biophys. Res. Commun. 2016, 470, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Yamaguchi, L.; Sharif, J.; Johmura, Y.; Kawamura, T.; Nakanishi, K.; Shimamura, S.; Arita, K.; Kodama, T.; Ishikawa, F.; et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 2013, 502, 249–253. [Google Scholar] [CrossRef]
- Qin, W.; Wolf, P.; Liu, N.; Link, S.; Smets, M.; La Mastra, F.; Forné, I.; Pichler, G.; Hörl, D.; Fellinger, K.; et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015, 25, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, L.; Nishiyama, A.; Misaki, T.; Johmura, Y.; Ueda, J.; Arita, K.; Nagao, K.; Obuse, C.; Nakanishi, M. Usp7-dependent histone H3 deubiquitylation regulates maintenance of DNA methylation. Sci. Rep. 2017, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Yarychkivska, O.; Tavana, O.; Gu, W.; Bestor, T.H. Independent functions of DNMT1 and USP7 at replication foci. Epigenet. Chromatin 2018, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Song, J.; Wang, Y.; Zhao, Y.; Guda, K.; Yang, S.; Kao, H.Y.; Xu, Y.; Willis, J.; Markowitz, S.D.; et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 2010, 3, ra80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, H.; Wang, Y.; Liu, W.; Zhang, Q.; Zhang, T.; Zhang, X.; Han, B.; Zhou, G. Epigenetic inactivation of the tumor suppressor gene RIZ1 in hepatocellular carcinoma involves both DNA methylation and histone modifications. J. Hepatol. 2010, 53, 889–895. [Google Scholar] [CrossRef]
- Izquierdo-Torres, E.; Hernández-Oliveras, A.; Meneses-Morales, I.; Rodríguez, G.; Fuentes-García, G.; Zarain-Herzberg, Á. Resveratrol up-regulates ATP2A3 gene expression in breast cancer cell lines through epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2019, 113, 37–47. [Google Scholar] [CrossRef]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Majid, S.; Dar, A.A.; Ahmad, A.E.; Hirata, H.; Kawakami, K.; Shahryari, V.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Khatri, G.; et al. BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 2009, 30, 662–670. [Google Scholar] [CrossRef]
- Kondo, Y.; Shen, L.; Issa, J.P. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol. Cell. Biol. 2003, 23, 206–215. [Google Scholar] [CrossRef]
- Jiao, D.; Huan, Y.; Zheng, J.; Wei, M.; Zheng, G.; Han, D.; Wu, J.; Xi, W.; Wei, F.; Yang, A.G.; et al. UHRF1 promotes renal cell carcinoma progression through epigenetic regulation of TXNIP. Oncogene 2019, 38, 5686–5699. [Google Scholar] [CrossRef]
- Beck, A.; Trippel, F.; Wagner, A.; Joppien, S.; Felle, M.; Vokuhl, C.; Schwarzmayr, T.; Strom, T.M.; von Schweinitz, D.; Längst, G.; et al. Overexpression of UHRF1 promotes silencing of tumor suppressor genes and predicts outcome in hepatoblastoma. Clin. Epigenet. 2018, 10, 27. [Google Scholar] [CrossRef]
- Shen, W.C.; Lai, Y.C.; Li, L.H.; Liao, K.; Lai, H.C.; Kao, S.Y.; Wang, J.; Chuong, C.M.; Hung, S.C. Methylation and PTEN activation in dental pulp mesenchymal stem cells promotes osteogenesis and reduces oncogenesis. Nat. Commun. 2019, 10, 2226. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Stewart, T.M.; Wu, Y.; Baylin, S.B.; Marton, L.J.; Perkins, B.; Jones, R.J.; Woster, P.M.; Casero, R.A., Jr. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7217–7228. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Chen, L.; Chen, Y.; Xu, S.G.; Di, G.H.; Yin, W.J.; Wu, J.; Shao, Z.M. UHRF1 is associated with epigenetic silencing of BRCA1 in sporadic breast cancer. Breast Cancer Res. Treat. 2010, 123, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Abusnina, A.; Keravis, T.; Yougbaré, I.; Bronner, C.; Lugnier, C. Anti-proliferative effect of curcumin on melanoma cells is mediated by PDE1A inhibition that regulates the epigenetic integrator UHRF1. Mol. Nutr. Food Res. 2011, 55, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Moseley, V.R.; Morris, J.; Knackstedt, R.W.; Wargovich, M.J. Green tea polyphenol epigallocatechin 3-gallate, contributes to the degradation of DNMT3A and HDAC3 in HCT 116 human colon cancer cells. Anticancer Res. 2013, 33, 5325–5333. [Google Scholar]
- Yu, C.; Xing, F.; Tang, Z.; Bronner, C.; Lu, X.; Di, J.; Zeng, S.; Liu, J. Anisomycin suppresses Jurkat T cell growth by the cell cycle-regulating proteins. Pharmacol. Rep. PR 2013, 65, 435–444. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, W.; Wang, Z.; Cai, P. Emodin promotes the arrest of human lymphoma Raji cell proliferation through the UHRF1‑DNMT3A‑∆Np73 pathways. Mol. Med. Rep. 2017, 16, 6544–6551. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Hong, D.; Jeong, S.Y.; Kim, J.H. Shikonin causes apoptosis by up-regulating p73 and down-regulating ICBP90 in human cancer cells. Biochem. Biophys. Res. Commun. 2015, 465, 71–76. [Google Scholar] [CrossRef]
- Krifa, M.; Leloup, L.; Ghedira, K.; Mousli, M.; Chekir-Ghedira, L. Luteolin induces apoptosis in BE colorectal cancer cells by downregulating calpain, UHRF1, and DNMT1 expressions. Nutr. Cancer 2014, 66, 1220–1227. [Google Scholar] [CrossRef]
- Krifa, M.; Alhosin, M.; Muller, C.D.; Gies, J.P.; Chekir-Ghedira, L.; Ghedira, K.; Mély, Y.; Bronner, C.; Mousli, M. Limoniastrum guyonianum aqueous gall extract induces apoptosis in human cervical cancer cells involving p16 INK4A re-expression related to UHRF1 and DNMT1 down-regulation. J. Exp. Clin. Cancer Res. CR 2013, 32, 30. [Google Scholar] [CrossRef]
- Jenkins, Y.; Markovtsov, V.; Lang, W.; Sharma, P.; Pearsall, D.; Warner, J.; Franci, C.; Huang, B.; Huang, J.; Yam, G.C.; et al. Critical role of the ubiquitin ligase activity of UHRF1, a nuclear RING finger protein, in tumor cell growth. Mol. Biol. Cell 2005, 16, 5621–5629. [Google Scholar] [CrossRef]
- Cheng, J.; Haas, M. Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Mol. Cell. Biol. 1990, 10, 5502–5509. [Google Scholar] [CrossRef]
- Shan, X.; Czar, M.J.; Bunnell, S.C.; Liu, P.; Liu, Y.; Schwartzberg, P.L.; Wange, R.L. Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol. Cell. Biol. 2000, 20, 6945–6957. [Google Scholar] [CrossRef]
- Shtraizent, N.; Matsui, H.; Polotskaia, A.; Bargonetti, J. Hot Spot Mutation in TP53 (R248Q) Causes Oncogenic Gain-of-Function Phenotypes in a Breast Cancer Cell Line Derived from an African American patient. Int. J. Environ. Res. Public Health 2015, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Nagel, J.H.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.S.; French, P.J.; Peeters, J.K.; Rozendaal, M.J.; et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat. 2010, 121, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Mousli, M.; Hopfner, R.; Abbady, A.Q.; Monté, D.; Jeanblanc, M.; Oudet, P.; Louis, B.; Bronner, C. ICBP90 belongs to a new family of proteins with an expression that is deregulated in cancer cells. Br. J. Cancer 2003, 89, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Jeanblanc, M.; Mousli, M.; Hopfner, R.; Bathami, K.; Martinet, N.; Abbady, A.Q.; Siffert, J.C.; Mathieu, E.; Muller, C.D.; Bronner, C. The retinoblastoma gene and its product are targeted by ICBP90: A key mechanism in the G1/S transition during the cell cycle. Oncogene 2005, 24, 7337–7345. [Google Scholar] [CrossRef] [PubMed]
- Gali-Muhtasib, H.U.; Abou Kheir, W.G.; Kheir, L.A.; Darwiche, N.; Crooks, P.A. Molecular pathway for thymoquinone-induced cell-cycle arrest and apoptosis in neoplastic keratinocytes. Anti-Cancer Drugs 2004, 15, 389–399. [Google Scholar] [CrossRef]
- Ivankovic, S.; Stojkovic, R.; Jukic, M.; Milos, M.; Milos, M.; Jurin, M. The antitumor activity of thymoquinone and thymohydroquinone in vitro and in vivo. Exp. Oncol. 2006, 28, 220–224. [Google Scholar]
- Shahein, S.A.; Aboul-Enein, A.M.; Higazy, I.M.; Abou-Elella, F.; Lojkowski, W.; Ahmed, E.R.; Mousa, S.A.; AbouAitah, K. Targeted anticancer potential against glioma cells of thymoquinone delivered by mesoporous silica core-shell nanoformulations with pH-dependent release. Int. J. Nanomed. 2019, 14, 5503–5526. [Google Scholar] [CrossRef]
- Gurung, R.L.; Lim, S.N.; Khaw, A.K.; Soon, J.F.; Shenoy, K.; Mohamed Ali, S.; Jayapal, M.; Sethu, S.; Baskar, R.; Hande, M.P. Thymoquinone induces telomere shortening, DNA damage and apoptosis in human glioblastoma cells. PLoS ONE 2010, 5, e12124. [Google Scholar] [CrossRef] [PubMed]
- Giri, A.K.; Aittokallio, T. DNMT Inhibitors Increase Methylation in the Cancer Genome. Front. Pharmacol. 2019, 10, 385. [Google Scholar] [CrossRef] [PubMed]
- Arafa el, S.A.; Zhu, Q.; Shah, Z.I.; Wani, G.; Barakat, B.M.; Racoma, I.; El-Mahdy, M.A.; Wani, A.A. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat. Res. 2011, 706, 28–35. [Google Scholar] [CrossRef]
- Şakalar, Ç.; İzgi, K.; İskender, B.; Sezen, S.; Aksu, H.; Çakır, M.; Kurt, B.; Turan, A.; Canatan, H. The combination of thymoquinone and paclitaxel shows anti-tumor activity through the interplay with apoptosis network in triple-negative breast cancer. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 4467–4477. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fei, C.; Zhang, X.; Zhang, Y.; Guo, J.; Gu, S.; Li, X.; Chang, C. Methylation of the p73 gene in patients with myelodysplastic syndromes: Correlations with apoptosis and prognosis. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2013, 34, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Chen, Y.C. Novel therapeutic targets for pancreatic cancer. World J. Gastroenterol. 2014, 20, 10825–10844. [Google Scholar] [CrossRef]
- Digel, W.; Lübbert, M. DNA methylation disturbances as novel therapeutic target in lung cancer: Preclinical and clinical results. Crit. Rev. Oncol. Hematol. 2005, 55, 1–11. [Google Scholar] [CrossRef]
- Wu, D.S.; Shen, J.Z.; Yu, A.F.; Fu, H.Y.; Zhou, H.R.; Shen, S.F. Epigallocatechin-3-gallate and trichostatin A synergistically inhibit human lymphoma cell proliferation through epigenetic modification of p16INK4a. Oncol. Rep. 2013, 30, 2969–2975. [Google Scholar] [CrossRef][Green Version]
- Mileo, A.M.; Di Venere, D.; Abbruzzese, C.; Miccadei, S. Long Term Exposure to Polyphenols of Artichoke (Cynara scolymus L.) Exerts Induction of Senescence Driven Growth Arrest in the MDA-MB231 Human Breast Cancer Cell Line. Oxidative Med. Cell. Longev. 2015, 2015, 363827. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Yan, Q.; Zhang, J.R.; Li, S.D.; Yang, Y.X.; Wan, X.P. Epigenetic inactivation of BRCA1 through promoter hypermethylation in ovarian cancer progression. J. Obstet. Gynaecol. Res. 2013, 39, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, L.; Lang, J. The BRCA1 Methylation and PD-L1 Expression in Sporadic Ovarian Cancer. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2018, 28, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Tokunaga, E.; Kitao, H.; Hitchins, M.; Inoue, Y.; Tanaka, K.; Hisamatsu, Y.; Taketani, K.; Akiyoshi, S.; Okada, S.; et al. Epigenetic Inactivation of BRCA1 Through Promoter Hypermethylation and Its Clinical Importance in Triple-Negative Breast Cancer. Clin. Breast Cancer 2015, 15, 498–504. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, J.; Sun, X.; Hao, M.; Ding, T.; Xiong, D.; Wang, X.; Zhu, Y.; Xiao, G.; Cheng, G.; et al. HIC1 modulates prostate cancer progression by epigenetic modification. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1400–1410. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, L.; Lin, J.; Huang, H.; Shi, B.; Lin, X.; Huang, Z.; Wang, C.; Qiu, J.; Wei, X. Hypermethylation of the HIC1 promoter and aberrant expression of HIC1/SIRT1 contribute to the development of thyroid papillary carcinoma. Oncotarget 2016, 7, 84416–84427. [Google Scholar] [CrossRef]
- Fleuriel, C.; Touka, M.; Boulay, G.; Guérardel, C.; Rood, B.R.; Leprince, D. HIC1 (Hypermethylated in Cancer 1) epigenetic silencing in tumors. Int. J. Biochem. Cell Biol. 2009, 41, 26–33. [Google Scholar] [CrossRef]
- Ghazanfari, T.; Asaadi Tehrani, G.; Maziri, P. The Relationship between the Methylation of Promoter Regions of Tumor Suppressor Genes PTEN and APC with Endometrial Cancer. Asian Pac. J. Cancer Prev. APJCP 2019, 20, 2259–2265. [Google Scholar] [CrossRef]
- Marsit, C.J.; Zheng, S.; Aldape, K.; Hinds, P.W.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. PTEN expression in non-small-cell lung cancer: Evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum. Pathol. 2005, 36, 768–776. [Google Scholar] [CrossRef]
- Osei-Amponsa, V.; Buckwalter, J.M.; Shuman, L.; Zheng, Z.; Yamashita, H.; Walter, V.; Wildermuth, T.; Ellis-Mohl, J.; Liu, C.; Warrick, J.I.; et al. Hypermethylation of FOXA1 and allelic loss of PTEN drive squamous differentiation and promote heterogeneity in bladder cancer. Oncogene 2020, 39, 1302–1317. [Google Scholar] [CrossRef]
- Bubna, A.K. Vorinostat-An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31. [Google Scholar] [CrossRef]
- Khot, A.; Dickinson, M.; Prince, H.M. Panobinostat in lymphoid and myeloid malignancies. Expert Opin. Investig. Drugs 2013, 22, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Bishton, M.J.; Johnstone, R.W. Panobinostat (LBH589): A potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009, 5, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Kumar, B.N.; Sarkar, S.; Das, S.; Azab, B.; Santhekadur, P.K.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B.; et al. Targeted apoptotic effects of thymoquinone and tamoxifen on XIAP mediated Akt regulation in breast cancer. PLoS ONE 2013, 8, e61342. [Google Scholar] [CrossRef] [PubMed]
- Ganji-Harsini, S.; Khazaei, M.; Rashidi, Z.; Ghanbari, A. Thymoquinone Could Increase The Efficacy of Tamoxifen Induced Apoptosis in Human Breast Cancer Cells: An In Vitro Study. Cell J. 2016, 18, 245–254. [Google Scholar]
- Singh, S.K.; Apata, T.; Gordetsky, J.B.; Singh, R. Docetaxel Combined with Thymoquinone Induces Apoptosis in Prostate Cancer Cells via Inhibition of the PI3K/AKT Signaling Pathway. Cancers 2019, 11, 1390. [Google Scholar] [CrossRef]
- Zafar, S.; Akhter, S.; Ahmad, I.; Hafeez, Z.; Alam Rizvi, M.M.; Jain, G.K.; Ahmad, F.J. Improved chemotherapeutic efficacy against resistant human breast cancer cells with co-delivery of Docetaxel and Thymoquinone by Chitosan grafted lipid nanocapsules: Formulation optimization, in vitro and in vivo studies. Colloids Surf. B Biointerfaces 2020, 186, 110603. [Google Scholar] [CrossRef]
- Hu, X.; Ma, J.; Vikash, V.; Li, J.; Wu, D.; Liu, Y.; Zhang, J.; Dong, W. Thymoquinone Augments Cisplatin-Induced Apoptosis on Esophageal Carcinoma Through Mitigating the Activation of JAK2/STAT3 Pathway. Dig. Dis. Sci. 2018, 63, 126–134. [Google Scholar] [CrossRef]
- Lei, X.; Lv, X.; Liu, M.; Yang, Z.; Ji, M.; Guo, X.; Dong, W. Thymoquinone inhibits growth and augments 5-fluorouracil-induced apoptosis in gastric cancer cells both in vitro and in vivo. Biochem. Biophys. Res. Commun. 2012, 417, 864–868. [Google Scholar] [CrossRef]
- Charles, M.R.C.; Mahesh, A.; Lin, S.Y.; Hsieh, H.P.; Dhayalan, A.; Coumar, M.S. Identification of novel quinoline inhibitor for EHMT2/G9a through virtual screening. Biochimie 2020, 168, 220–230. [Google Scholar] [CrossRef]
- Cao, Y.P.; Sun, J.Y.; Li, M.Q.; Dong, Y.; Zhang, Y.H.; Yan, J.; Huang, R.M.; Yan, X. Inhibition of G9a by a small molecule inhibitor, UNC0642, induces apoptosis of human bladder cancer cells. Acta Pharmacol. Sin. 2019, 40, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, J.; Yang, L.; Yuan, Y.C.; Tong, T.R.; Wu, J.; Yun, X.; Bonner, M.; Pangeni, R.; Liu, Z.; et al. Targeting histone methyltransferase G9a inhibits growth and Wnt signaling pathway by epigenetically regulating HP1α and APC2 gene expression in non-small cell lung cancer. Mol. Cancer 2018, 17, 153. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epi-Target | Role of Epi-Target | Experimental Model | Mechanisms of Action | References |
|---|---|---|---|---|
| UHRF1 | Reader | Human cervical carcinoma HeLa cells. T-ALL | TQ targeted the E3 ubiquitin ligase activity of UHRF1 resulting in an auto-ubiquitination of UHRF1 likely through the downregulation of HAUSP | [23] |
| T-ALL | TQ upregulated p73 expression and cleaved caspase 3 leading to UHRF1 degradation | [63] | ||
| T-ALL | TQ decreased the expression of PDE1A leading to the upregulation of p73 and downregulation of UHRF1 | [62] | ||
| T-ALL Human breast cancer cells | TQ decreased the expression of mRNA UHRF1 in dose-dependent mechanism | [60] | ||
| DNMT1 DNMT3A DNMT3B | Writer | Human acute myeloid leukemia cells Patient primary cells | TQ inhibited DNMT1 activity and decreased its expression through the disruption of Sp1/NFkB complex from DNMT1 promoter. TQ decreased the expression of DNMT3A through the upregulation of miR-29b, known to directly bind to the 3′-UTR of DNMT3A | [64] |
| T-ALL | TQ decreased the expression of DNMT1 protein | [63] | ||
| T-ALL | TQ decreased the expression of DNMT1, 3A,3B | [60] | ||
| HDAC1 HDAC2 HDAC3 HDAC4 HDAC9 | Eraser | T-ALL | TQ decreased the expression of HDAC1 protein | [63] |
| T-ALL | TQ decreased in the expression of HDAC1, 4 and 9 | [60] | ||
| T-ALL Human breast cancer cells | TQ decreased the expression of mRNA HDAC1 in dose-dependent mechanism | [60] | ||
| Human pancreatic ductal adenocarcinoma cells. Human pancreatic ductal adenocarcinoma xenografts. | TQ inhibited HDAC activity, decreased the expression of HDAC 1, 2, 3 at mRNA levels and increased the acetylation of histone 4 at lysine 12 (H4 Ac-K12) | [65] | ||
| G9A | Writer | T-ALL Human breast cancer cells | TQ decreased the expression of mRNA G9A in dose-dependent mechanism | [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah, O.; Omran, Z.; Hosawi, S.; Hamiche, A.; Bronner, C.; Alhosin, M. Thymoquinone Is a Multitarget Single Epidrug That Inhibits the UHRF1 Protein Complex. Genes 2021, 12, 622. https://doi.org/10.3390/genes12050622
Abdullah O, Omran Z, Hosawi S, Hamiche A, Bronner C, Alhosin M. Thymoquinone Is a Multitarget Single Epidrug That Inhibits the UHRF1 Protein Complex. Genes. 2021; 12(5):622. https://doi.org/10.3390/genes12050622
Chicago/Turabian StyleAbdullah, Omeima, Ziad Omran, Salman Hosawi, Ali Hamiche, Christian Bronner, and Mahmoud Alhosin. 2021. "Thymoquinone Is a Multitarget Single Epidrug That Inhibits the UHRF1 Protein Complex" Genes 12, no. 5: 622. https://doi.org/10.3390/genes12050622
APA StyleAbdullah, O., Omran, Z., Hosawi, S., Hamiche, A., Bronner, C., & Alhosin, M. (2021). Thymoquinone Is a Multitarget Single Epidrug That Inhibits the UHRF1 Protein Complex. Genes, 12(5), 622. https://doi.org/10.3390/genes12050622