
Molecular Phylogeny and Evolution of Amazon Parrots in the Greater Antilles

 , , , and
, , , and 
Abstract
1. Introduction

2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. De Novo Mitogenome Sequencing and Assembly
2.3. Reference-Based Mitogenome Assemblies
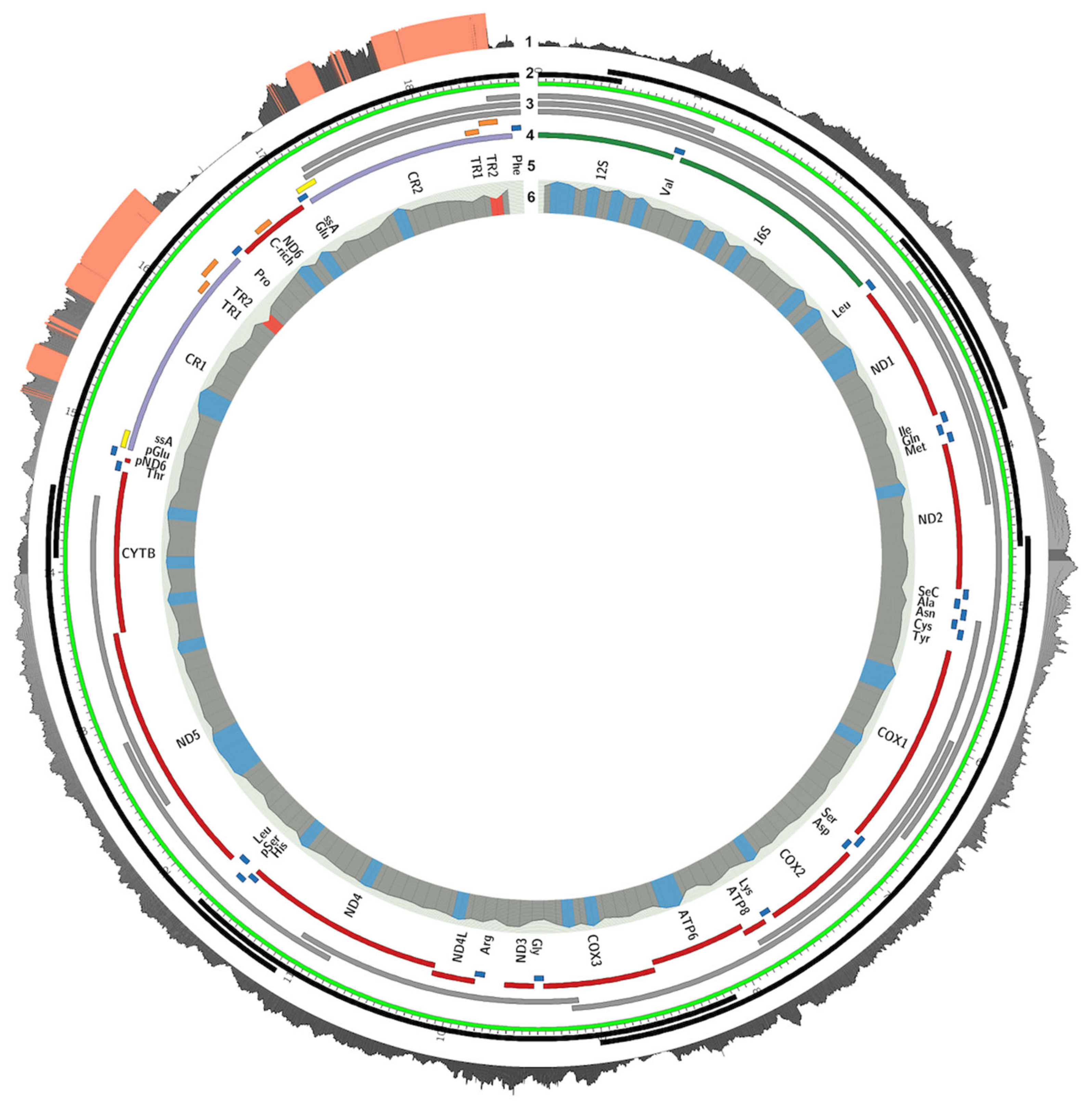
2.4. Mitogenome Annotation
2.5. Mitogenome Sequence Capture for Annotating Diversity within Species
2.6. Multiple Alignments and Phylogenetic Analyses
2.7. Reconstructing Dispersal and Speciation
3. Results
3.1. Mitogenome Sequencing, Assembly, and Annotation
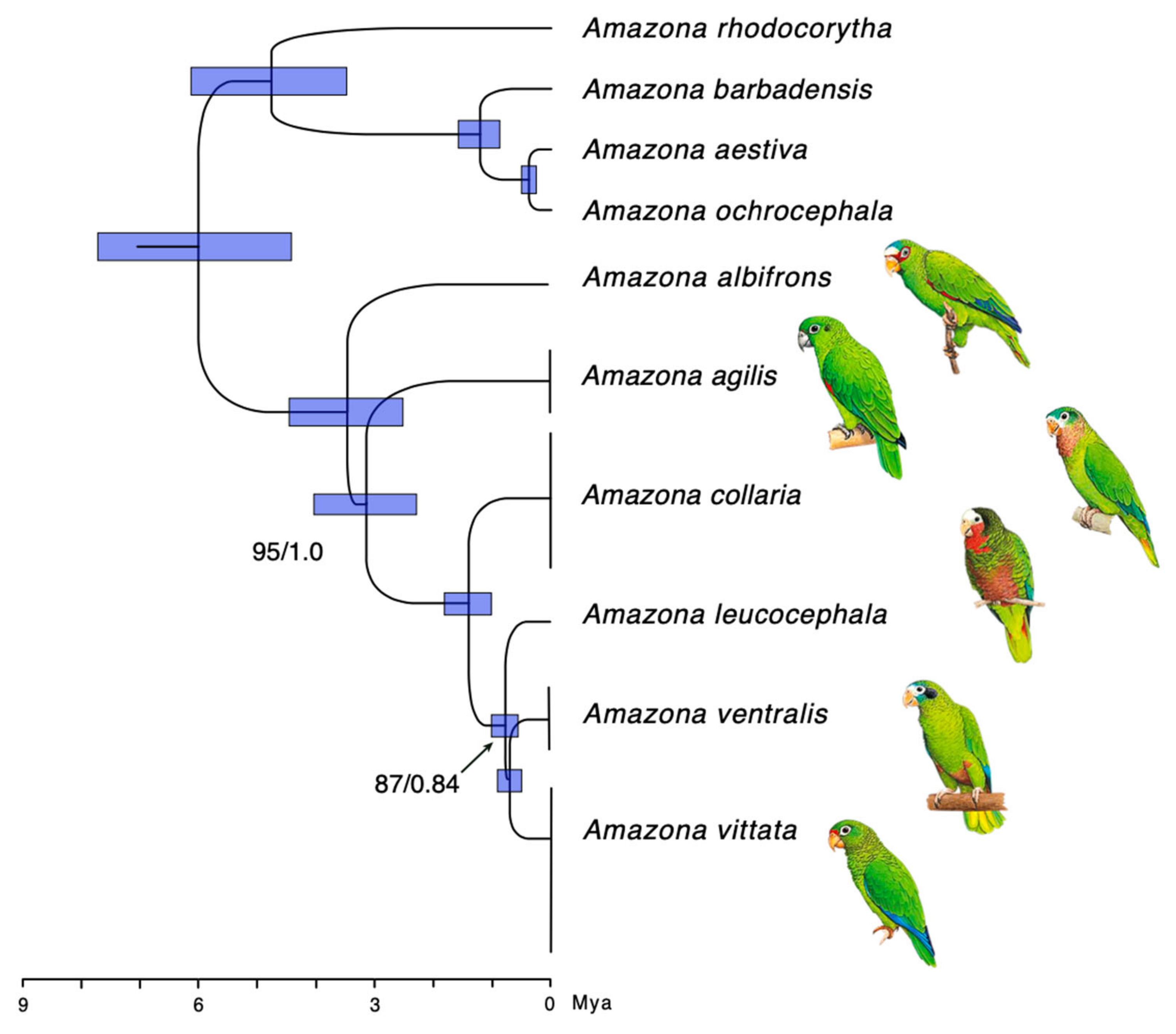
3.2. Phylogeny and Evolutionary History
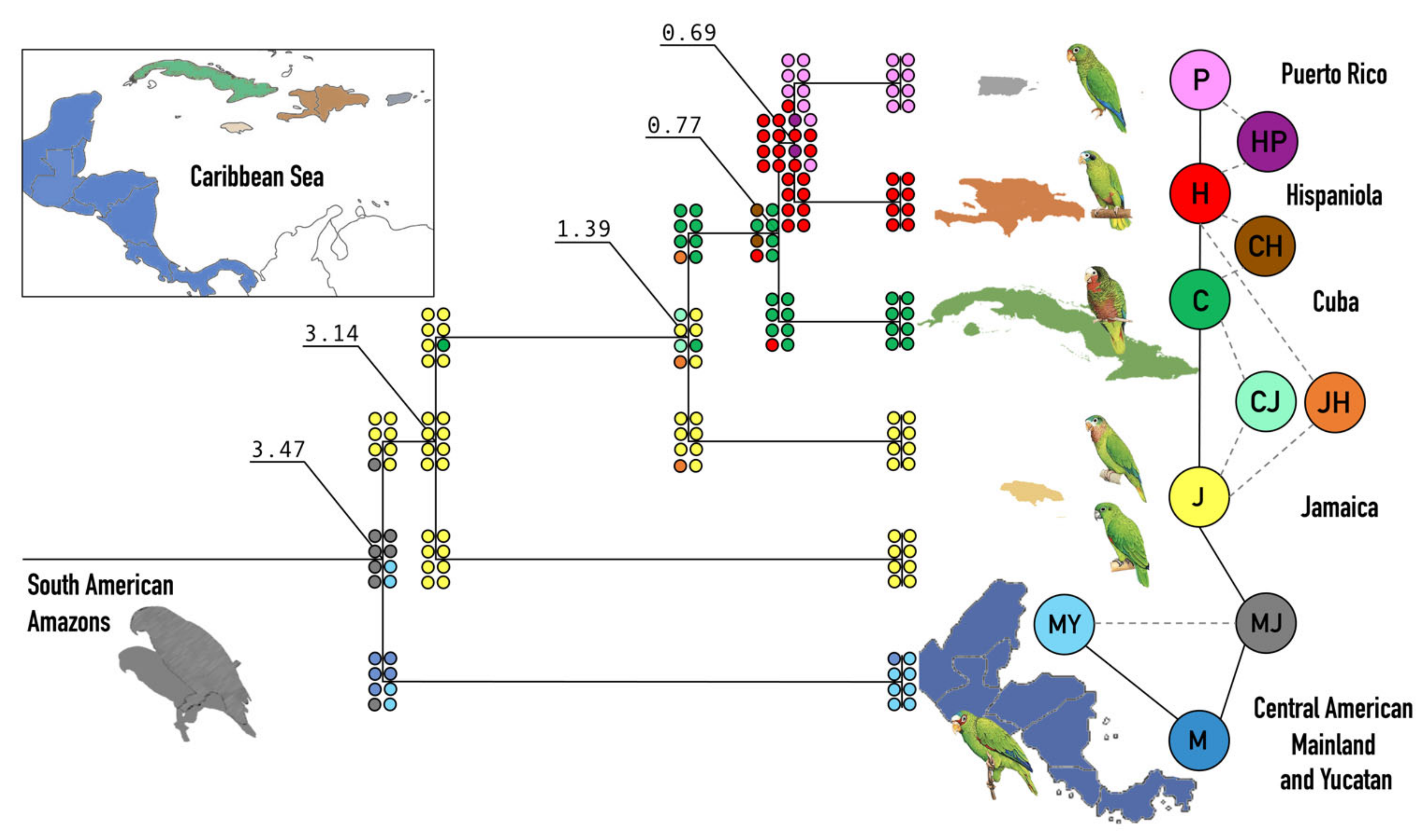
3.3. Dispersal and Speciation
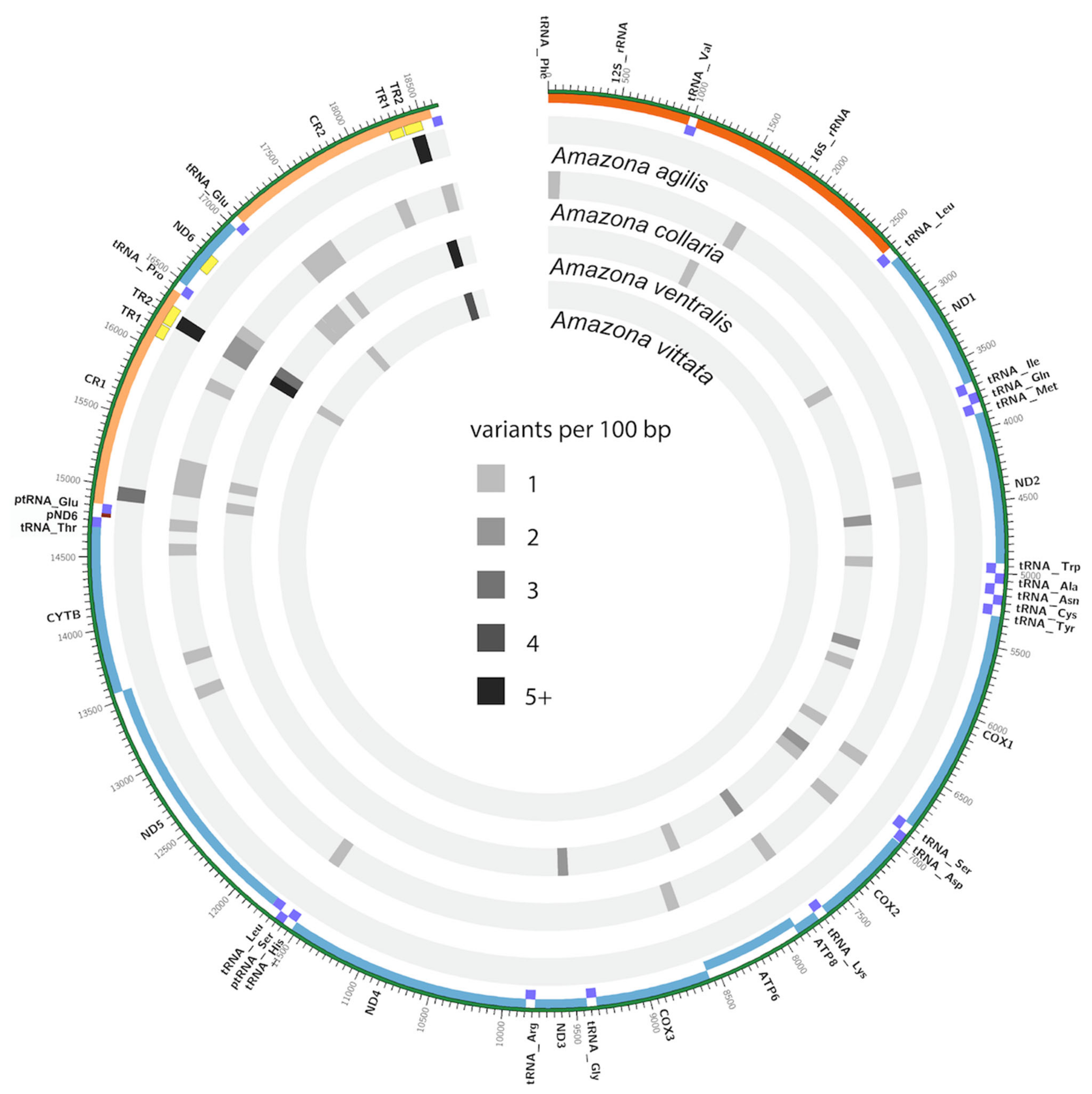
3.4. Mitogenome Diversity
4. Discussion
4.1. Mitogenome Diversity
4.2. Phylogeny
4.3. Biogeography
4.4. Conservation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
Appendix C
References
- Darwin, C. A Naturalist’s Voyage Round the World/ The Voyage of The Beagle, 11th ed.; John Murray: London, UK, 1860. [Google Scholar]
- MacArthur, R.H.; Wilson, E.O. The Theory of Island Biogeography; Princeton University Press: Princeton, NJ, USA, 1967; ISBN 9781400875368. [Google Scholar]
- Grant, P.R.; Grant, B.R. 40 Years of Evolution; Princeton University Press: Princeton, NJ, USA, 2014; ISBN 9781400851300. [Google Scholar]
- Whittaker, R.J.; Fernández-Palacios, J.M.; Matthews, T.J.; Borregaard, M.K.; Triantis, K.A. Island Biogeography: Taking the Long View of Natures Laboratories. Science 2017, 357. [Google Scholar] [CrossRef]
- Losos, J.B.; Ricklefs, R.E. Adaptation and Diversification on Islands. Nature 2009, 457, 830–836. [Google Scholar] [CrossRef]
- Losos, J.B.; Ricklefs, R.E. The Theory of Island Biogeography Revisited; Princeton University Press: Princeton, NJ, USA, 2010; ISBN 9781400831920. [Google Scholar]
- Diamond, J.M. The Present, Past and Future of Human-Caused Extinctions. Biol. Sci. 1989, 325, 469–476. [Google Scholar] [CrossRef]
- Bennett, P.M.; Owens, I.P.F. Variation in Extinction Risk among Birds: Chance or Evolutionary Predisposition? Proc. R. Soc. B Biol. Sci. 1997, 264, 401–408. [Google Scholar] [CrossRef]
- Butchart, S.H.M.; Stattersfield, A.J.; Bennun, L.A.; Shutes, S.M.; Akçakaya, H.R.; Baillie, J.E.M.; Stuart, S.N.; Hilton-Taylor, C.; Mace, G.M. Measuring Global Trends in the Status of Biodiversity: Red List Indices for Birds. PLoS Biol. 2004, 2. [Google Scholar] [CrossRef]
- Forshaw, J.M. Vanished and Vanishing Parrots; CSIRO Publishing: Melbourne, Australia, 2017. [Google Scholar]
- Toft, C.A.; Wright, T.F. Parrots of the Wild. A Natural History of the World’s Most Captivating Birds; University of California Press: Oakland, CA, USA, 2015. [Google Scholar]
- IUCN. The IUCN Red List of Threatened Species. 2021. Available online: https://www.iucnredlist.org/ (accessed on 12 April 2021).
- Berkunsky, I.; Quillfeldt, P.; Brightsmith, D.J.; Abbud, M.C.; Aguilar, J.M.R.E.; Alemán-Zelaya, U.; Aramburú, R.M.; Arce Arias, A.; Balas McNab, R.; Balsby, T.J.S.; et al. Current Threats Faced by Neotropical Parrot Populations. Biol. Conserv. 2017, 214, 278–287. [Google Scholar] [CrossRef]
- Wiley, J.; Gnam, R.; Koenig, S.; Dornelly, A.; Gálvez, X.; Bradley, P.; White Thomas, J.; Zamore, M.; Reillo, R.P. Status and Conservation of the Family Psittacidae in the West Indies. J. Caribb. Ornithol. 2004, 17, 94–154. [Google Scholar]
- Snyder, N.F.R.; Wiley, J.W.; Kepler, C.B. The Parrots of Luquillo: Natural History and Conservation of the Puerto Rican Parrot; Western Foundation of Vertebrate Zoology: Los Angeles, CA, USA, 1987. [Google Scholar]
- Russello, M.A.; Amato, G. A Molecular Phylogeny of Amazona: Implications for Neotropical Parrot Biogeography, Taxonomy, and Conservation. Mol. Phylogenetics Evol. 2004, 30, 421–437. [Google Scholar] [CrossRef]
- Lack, D. Island Biology, Illustrated by the Land Birds of Jamaica; University of California Press: Berkeley, CA, USA, 1976. [Google Scholar]
- Bond, J. Birds of the West Indies, 5th ed.; The Peterson Field Guide Series; Houghton Mifflin: Boston, MA, USA, 1971; ISBN 9780395074312. [Google Scholar]
- Lantermann, W. Verbreitung Und Evolution Der Psittacidenfauna Auf Den Ozeanischen Inseln Der Karibischen See. Papageienkunde Parrot Biol. 1997, 1, 263–278. [Google Scholar]
- Ottens-Wainright, P.; Halanych, K.M.; Eberhard, J.R.; Burke, R.I.; Wiley, J.W.; Gnam, R.S.; Aquilera, X.G. Independent Geographic Origin of the Genus Amazona in the West Indies. J. Caribb. Ornithol. 2004, 17, 23–49. [Google Scholar]
- Provost, K.L.; Joseph, L.; Smith, B.T. Resolving a Phylogenetic Hypothesis for Parrots: Implications from Systematics to Conservation. Emu Austral Ornithology 2018, 118, 7–21. [Google Scholar] [CrossRef]
- Armstrong, G. Conservation and the Genetics of Populations; John Wiley & Sons: Hoboken, NJ, USA, 2008; Volume 14. [Google Scholar]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the Future of Conservation Genetics. Nat. Reviews. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Kolchanova, S.; Kliver, S.; Komissarov, A.; Dobrinin, P.; Tamazian, G.; Grigorev, K.; Wolfsberger, W.W.; Majeske, A.J.; Velez-Valentin, J.; de la Rosa, R.V.; et al. Genomes of Three Closely Related Caribbean Amazons Provide Insight for Species History and Conservation. Genes 2019, 10, 54. [Google Scholar] [CrossRef]
- Oleksyk, T.K.; Pombert, J.F.; Siu, D.; Mazo-Vargas, A.; Ramos, B.; Guiblet, W.; Afanador, Y.; Ruiz-Rodriguez, C.T.; Nickerson, M.L.; Logue, D.M.; et al. A Locally Funded Puerto Rican Parrot (Amazona Vittata) Genome Sequencing Project Increases Avian Data and Advances Young Researcher Education. GigaScience 2012, 1, 14. [Google Scholar] [CrossRef]
- Urantowka, A.D.; Hajduk, K.; Kosowska, B. Complete Mitochondrial Genome of Endangered Yellow-Shouldered Amazon (Amazona Barbadensis): Two Control Region Copies in Parrot Species of the Amazona Genus. Mitochondrial DNA 2013, 24, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and Efficient Long Read Error Correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Starostina, E.; Tamazian, G.; Dobrynin, P.; O’Brien, S.; Komissarov, A. Cookiecutter: A Tool for Kmer-Based Read Filtering and Extraction, version 1.0.0. BioRxiv 2015. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Marcais, G.; Kingsford, C. Jellyfish: A Fast k-Mer Counter, version 1.1.4. Tutor. E Manuais: 2012. Available online: http://www.cbcb.umd.edu/software/jellyfish/ (accessed on 5 April 2019).
- Chaisson, M.J.; Tesler, G. Mapping Single Molecule Sequencing Reads Using Basic Local Alignment with Successive Refinement (BLASR): Application and Theory. Bmc Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef]
- Urantowka, A.D.; Kroczak, A.; Mackiewicz, P. Complete Mitochondrial Genome of the Greater Antillean Parrot Amazona Ventralis (Hispaniolan Amazon). Mitochondrial DNA Part B 2016, 1, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Hains, T.; O’Neill, K.; Velez, J.; Speed, N.; Clubb, S.; Oleksyk, T.; Pirro, S. The Complete Genome Sequences of 22 Parrot Species (Psittaciformes, Aves) [Version 1; Peer Review: 1 Approved with Reservations]. F1000Research 2020, 9, 1318. [Google Scholar] [CrossRef]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and Quality Management in Next-Generation Sequencing. Appl. Transl. Genom. 2016, 10, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct Determination of Diploid Genome Sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High Speed BLASTN: An Accelerated MegaBLAST Search Tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. TRNAscan-SE On-Line: Integrating Search and Context for Analysis of Transfer RNA Genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.V.; Yuhki, N.; Masuda, R.; Modi, W.; O’Brien, S.J. Numt, a Recent Transfer and Tandem Amplification of Mitochondrial DNA to the Nuclear Genome of the Domestic Cat. J. Mol. Evol. 1994, 39, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Nacer, D.F.; Raposo do Amaral, F. Striking Pseudogenization in Avian Phylogenetics: Numts Are Large and Common in Falcons. Mol. Phylogenet. Evol. 2017, 115, 1–6. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- MyBaits. Hybridization Capture for Targeted NGS—Manual, version 5.01. 2021. Available online: www.arborbiosci.com/mybaits-manual (accessed on 5 April 2019).
- Lassmann, T. Kalign 3: Multiple Sequence Alignment of Large Datasets. Bioinformatics 2019, 36, 1928–1929. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Ugene Team. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for MacOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partitionfinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D. JModelTest 2.0 Manual, version 0.1.10. 2012. Available online: https://github.com/ddarriba/jmodeltest2 (accessed on 5 April 2019).
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nature Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree, version 1.4.3. 2016. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 8 November 2018).
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. Plos Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.R.; Drummond, A.J. BModelTest: Bayesian Phylogenetic Site Model Averaging and Model Comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef]
- Rheindt, F.E.; Christidis, L.; Kuhn, S.; de Kloet, S.; Norman, J.A.; Fidler, A. The Timing of Diversification within the Most Divergent Parrot Clade. J. Avian Biol. 2014, 45, 140–148. [Google Scholar] [CrossRef]
- Jetz, W.; Thomas, G.H.; Joy, J.B.; Hartmann, K.; Mooers, A.O. The Global Diversity of Birds in Space and Time. Nature 2012. [Google Scholar] [CrossRef]
- Quintero, E.; Ribas, C.C.; Cracraft, J. The Andean Hapalopsittaca Parrots (Psittacidae, Aves): An Example of Montane-Tropical Lowland Vicariance. Zool. Scr. 2013, 42, 28–43. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. Tracer: MCMC Trace Analysis Tool; University of Oxford: Oxford, UK, 2009. [Google Scholar]
- Li, W.L.S.; Drummond, A.J. Model Averaging and Bayes Factor Calculation of Relaxed Molecular Clocks in Bayesian Phylogenetics. Mol. Biol. Evol. 2012, 29, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Matzke, N.J. BioGeoBEARS: BioGeography with Bayesian (and Likelihood) Evolutionary Analysis in R Scripts, R Package version 0.2; University of California: Berkeley, CA, USA, 2013. Available online: http://phylo.wikidot.com/biogeobears (accessed on 20 September 2020).
- Matzke, N.J. Model Selection in Historical Biogeography Reveals That Founder-Event Speciation Is a Crucial Process in Island Clades. Syst. Biol. 2014, 63, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F. Ancestral Areas and Parsimony. Syst. Biol. 1994, 43, 267–274. [Google Scholar] [CrossRef]
- Ronquist, F. Phylogenetic Methods in Historical Biogeography. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 441–464. [Google Scholar] [CrossRef]
- Ree, R.H. Detecting the Historical Signature of Key Innovations Using Stochastic Models of Character Evolution and Cladogenesis. Evolution 2005, 59, 257–265. [Google Scholar] [CrossRef]
- Ree, R.H.; Smith, S.A. Maximum Likelihood Inference of Geographic Range Evolution by Dispersal, Local Extinction, and Cladogenesis. Syst. Biol. 2008, 57, 4–14. [Google Scholar] [CrossRef]
- Landis, M.J.; Matzke, N.J.; Moore, B.R.; Huelsenbeck, J.P. Bayesian Analysis of Biogeography When the Number of Areas Is Large. Syst. Biol. 2013, 62, 789–804. [Google Scholar] [CrossRef]
- Ronquist, F. DIVA 1.1 User’s Manual, version 1.1; Uppsala University: Uppsala, Sweden, 1996. Available online: http://ib.berkeley.edu/courses/ib200b/labs/lab15/divawin/ (accessed on 20 September 2020).
- Ree, R.H.; Moore, B.R.; Webb, C.O.; Donoghue, M.J. A Likelihood Framework for Inferring the Evolution of Geographic Range on Phylogenetic Trees. Evolution 2005, 59, 2299–2311. [Google Scholar] [CrossRef]
- Matzke, N.J. Probabilistic Historical Biogeography: New Models for Founder-Event Speciation, Imperfect Detection, and Fossils Allow Improved Accuracy and Model-Testing. Front. Biogeogr. 2013, 5. [Google Scholar] [CrossRef]
- Donnely, T. Geologic constraints on Caribbean biogeography. In Zoogeography of Caribbean Insects; Liebherr, J.K., Ed.; Cornell University Press: Ithaca, NY, USA, 1988; pp. 15–37. [Google Scholar]
- Hedges, S.B. Biogeography of the West Indies: An Overview. In Biogeography of the West Indies: Patterns and Perspectives; Charles, A., Woods, F.E.S., Eds.; CRC Press: Boca Raton, FL, USA, 2001; pp. 15–33. [Google Scholar]
- Purdue, J.R.; Oleksyk, T.K.; Smith, M.H. Independent Occurrences of Multiple Repeats in the Control Region of Mitochondrial DNA of White-Tailed Deer. J. Hered. 2006, 97, 235–243. [Google Scholar] [CrossRef][Green Version]
- Eberhard, J.R.; Wright, T.F. Rearrangement and Evolution of Mitochondrial Genomes in Parrots. Mol. Phylogenet. Evol. 2016, 94, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Lima, N.C.B.; Soares, A.E.R.; de Almeida, L.G.P.; da Costa, I.R.; Sato, F.M.; Schneider, P.; Aleixo, A.; Schneider, M.P.; Santos, F.R.; Mello, C.V.; et al. Comparative Mitogenomic Analyses of Amazona Parrots and Psittaciformes. Genet. Mol. Biol. 2018, 41, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Wang, N.; Li, N.; Kimball, R.T.; Braun, E.L. Comparative Genomics Reveals a Burst of Homoplasy-Free Numt Insertions. Mol. Biol. Evol. 2018, 35, 2060–2064. [Google Scholar] [CrossRef] [PubMed]
- Kolchanova, S. Molecular Phylogeny and Evolution of Amazon Parrots in the Greater Antilles; Oleksyk, T.K., Ed.; University of Puerto Rico at Mayaguez: Mayaguez, Puerto Rico, 2018. [Google Scholar]
- Kitchener, A. How and Why Species Multiply. The Radiation of Darwin’s Finches. Biol. J. Linn. Soc. 2008, 95, 653–654. [Google Scholar] [CrossRef]
- O’Brien, S.J. Genome Empowerment for the Puerto Rican Parrot—Amazona vittata. GigaScience 2012, 1, 13. [Google Scholar] [CrossRef]
- Oleksyk, T.K.; Smith, M.W.; O’Brien, S.J. Genome-Wide Scans for Footprints of Natural Selection. Philos. Trans. R. Soc. London. Ser. Bbiolog. Sci. 2010, 365, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative Genomics Reveals Insights into Avian Genome Evolution and Adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-Genome Analyses Resolve Early Branches in the Tree of Life of Modern Birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Whitlock, M.C. The Incomplete Natural History of Mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, P.; Urantówka, A.D.; Kroczak, A.; Mackiewicz, D. Resolving Phylogenetic Relationships within Passeriformes Based on Mitochondrial Genes and Inferring the Evolution of Their Mitogenomes in Terms of Duplications. Genome Biol. Evol. 2019, 11, 2824–2849. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B. Vicariance and Dispersal in Caribbean Biogeography. Herpetologica 1996, 52, 466–473. [Google Scholar] [CrossRef]
- Boschman, L.M.; van Hinsbergen, D.J.J.; Torsvik, T.H.; Spakman, W.; Pindell, J.L. Kinematic Reconstruction of the Caribbean Region since the Early Jurassic. Earth Sci. Rev. 2014, 138, 102–136. [Google Scholar] [CrossRef]
- Montes, C.; Cardona, A.; Jaramillo, C.; Pardo, A.; Silva, J.C.; Valencia, V.; Ayala, C.; Pérez-Angel, L.C.; Rodriguez-Parra, L.A.; Ramirez, V.; et al. Middle Miocene Closure of the Central American Seaway. Science 2015, 348, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Wadge, G.; Dixon, T.H. A Geological Interpretation of Seasat-Sar Imagery of Jamaica. J. Geol. 1984, 92, 561–581. [Google Scholar] [CrossRef]
- O’Dea, A.; Lessios, H.A.; Coates, A.G.; Eytan, R.I.; Restrepo-Moreno, S.A.; Cione, A.L.; Collins, L.S.; De Queiroz, A.; Farris, D.W.; Norris, R.D.; et al. Formation of the Isthmus of Panama. Sci. Adv. 2016, 2, e1600883. [Google Scholar] [CrossRef]
- Leigh, E.G.; O’Dea, A.; Vermeij, G.J. Historical Biogeography of the Isthmus of Panama. Biol. Rev. 2014, 89, 148–172. [Google Scholar] [CrossRef]
- Mutti, M.; Droxler, A.W.; Cunningham, A.D. Evolution of the Northern Nicaragua Rise during the Oligocene-Miocene: Drowning by Environmental Factors. Sediment. Geol. 2005, 175, 237–258. [Google Scholar] [CrossRef]
- Hedges, S.B. Historical Biogeography of West Indian Vertebrates. Annu. Rev. Ecol. Syst. 1996, 27, 163–196. [Google Scholar] [CrossRef]
- Iturralde-Vinent, M.A.; MacPhee, R.D.E. Paleogeography of the Caribbean Region: Implications for Cenozoic Biogeography. Bulletin AMNH 1999, 238, 1–95. [Google Scholar]
- Iturralde-Vinent, M.A. Meso-Cenozoic Caribbean Paleogeography: Implications for the Historical Biogeography of the Region. Int. Geol. Rev. 2006, 48, 791–827. [Google Scholar] [CrossRef]
- Bacon, C.D.; Silvestro, D.; Jaramillo, C.; Smith, B.T.; Chakrabarty, P.; Antonelli, A. Biological Evidence Supports an Early and Complex Emergence of the Isthmus of Panama. Proc. Natl. Acad. Sci. USA 2015, 112, 6110–6115. [Google Scholar] [CrossRef]
- Grigorev, K.; Kliver, S.; Dobrynin, P.; Komissarov, A.; Wolfsberger, W.; Krasheninnikova, K.; Afanador-Hernández, Y.M.; Brandt, A.L.; Paulino, L.A.; Carreras, R.; et al. Innovative Assembly Strategy Contributes to Understanding the Evolution and Conservation Genetics of the Endangered Solenodon Paradoxus from the Island of Hispaniola. GigaScience 2018, 6, giy025. [Google Scholar] [CrossRef]
- Brandt, A.L.; Grigorev, K.; Afanador-Hernández, Y.M.; Paulino, L.A.; Murphy, W.J.; Núñez, A.; Komissarov, A.; Brandt, J.R.; Dobrynin, P.; Hernández-Martich, J.D.; et al. Mitogenomic Sequences Support a North–South Subspecies Subdivision within Solenodon Paradoxus. Mitochondrial DNA Part A Dna Mapp. Seq. Anal. 2017, 28, 662–670. [Google Scholar] [CrossRef]
- Díaz-Lameiro, A.M.; Oleksyk, T.K.; Bird-Picó, F.J.; Martínez-Cruzado, J.C. Colonization of Islands in the Mona Passage by Endemic Dwarf Geckoes (Genus Sphaerodactylus) Reconstructed with Mitochondrial Phylogeny. Ecol. Evol. 2013, 3, 4488–4500. [Google Scholar] [CrossRef] [PubMed]
- Censky, E.J.; Hodge, K.; Dudley, J. Over-Water Dispersal of Lizards Due to Hurricanes. Nature 1998, 395, 556. [Google Scholar] [CrossRef]
- Whittaker, R.J.; Fernandez-Palacios, J.M. Island Biogeography: Ecology, Evolution, and Conservation; Oxford Biology Island Biogeography; OUP Oxford: Oxford, UK, 2007. [Google Scholar]
- Nunn, P.D. Oceanic Islands (Natural Environment); Blackwell: Oxford, UK; Cambridge, MA, USA, 1994. [Google Scholar]
- Coyne, J.A.; Price, T.D. Little evidence for sympatric speciation in island birds. Evolution 2000, 54, 2166–2171. [Google Scholar] [CrossRef]
- Phillimore, A.B.; Orme, C.D.L.; Thomas, G.H.; Blackburn, T.M.; Bennett, P.M.; Gaston, K.J.; Owens, I.P.F. Sympatric Speciation in Birds Is Rare: Insights from Range Data and Simulations. Am. Nat. 2008, 171, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Collar, N.; Boesman, P.F.D.; Kirwan, G.M.; Sharpe, C.J. Cuban Parrot (Amazona leucocephala). In Birds of the World; del Hoyo, J.A., Elliott, A., Sargatal, J., Christie, D.A., de Juana, E., Eds.; Cornell Lab of Ornithology: Ithaca, NY, USA, 2020. [Google Scholar]
- Donovan, S.K.; Jackson, T.A. Caribbean Geology: An Introduction; University of the West Indies Publishers’ Association: West Indies, Jamaica, 1994. [Google Scholar]
- Eizaguirre, C.; Baltazar-Soares, M. Evolutionary Conservation—Evaluating the Adaptive Potential of Species. Evol. Appl. 2014, 7, 963–967. [Google Scholar] [CrossRef]
- Brandies, P.; Peel, E.; Hogg, C.J.; Belov, K. The Value of Reference Genomes in the Conservation of Threatened Species. Genes 2019, 10, 846. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, I.; Tonnabel, J.; Ronce, O.; Mignot, A. Why Evolution Matters for Species Conservation: Perspectives from Three Case Studies of Plant Metapopulations. Evol. Appl. 2016, 9, 196–211. [Google Scholar] [CrossRef]
- Rosauer, D.F.; Pollock, L.J.; Linke, S.; Jetz, W. Phylogenetically Informed Spatial Planning Is Required to Conserve the Mammalian Tree of Life. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170627. [Google Scholar] [CrossRef]
- Crandall, K.A.; Bininda-Emonds, O.R.P.; Mace, G.M.; Wayne, R.K. Considering Evolutionary Processes in Conservation Biology. Trends Ecol. Evol. 2000, 15, 290–295. [Google Scholar] [CrossRef]
- Hartl, D.L.; Conner, J.K. A Primer of Ecological Genetics; Sinauer: Sunderland, MA, USA, 2004; ISBN 087893202X. [Google Scholar]
- Johnson, W.E.; Onorato, D.P.; Roelke, M.E.; Land, E.D.; Cunningham, M.; Belden, R.C.; McBride, R.; Jansen, D.; Lotz, M.; Shindle, D.; et al. Genetic Restoration of the Florida Panther. Science 2010, 329, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W. Gene Flow and Genetic Restoration: The Florida Panther as a Case Study. Conserv. Biol. 1995, 9, 996–1007. [Google Scholar] [CrossRef]
- Macarthur, R.H.; Wilson, E. An Equilibrium Theory of Insular Zoogeography. Evolution 1963, 17, 373–387. [Google Scholar] [CrossRef]
- BirdLife International IUCN Red List for Birds. 2021. Available online: https://www.birdlife.org/redlist (accessed on 1 March 2021).
- Nei, M.; Li, W.H. Mathematical Model for Studying Genetic Variation in Terms of Restriction Endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Berlin, S.; Tomaras, D.; Charlesworth, B. Low Mitochondrial Variability in Birds May Indicate Hill–Robertson Effects on the W Chromosome. Heredity 2007, 99, 389–396. [Google Scholar] [CrossRef]
- Koenig, S.E. The Breeding Biology of Black-Billed Parrot Amazona agilis and Yellow-Billed Parrot Amazona collaria in Cockpit Country, Jamaica. Bird Conserv. Int. 2001, 11, 205–225. [Google Scholar] [CrossRef]
- Koenig, S.E.; Wunderle, J.M.; Enkerlin-Hoeflich, E.C. Vines and Canopy Contact: A Route for Snake Predation on Parrot Nests. Bird Conserv. Int. 2007, 17, 79–91. [Google Scholar] [CrossRef]
- Newton, I. The Role of Nest Sites in Limiting the Numbers of Hole-Nesting Birds: A Review. Biol. Conserv. 1994, 70, 265–276. [Google Scholar] [CrossRef]
- Wiebe, K.L. Nest Sites as Limiting Resources for Cavity-Nesting Birds in Mature Forest Ecosystems: A Review of the Evidence. J. Field Ornithol. 2011, 82, 239–248. [Google Scholar] [CrossRef]
- Cruz, A.; Gruber, S. The distribution, ecology, and breeding biology of Jamaican amazon parrots. In Conservation of the New World parrots, Proceedings of the ICBP (International Council for Bird Preservation) Parrot Working Group Meeting, St. Lucia, 1980; ICBP (International Council for Bird Preservation) IUCNWWF; Smithsonian Institution Press: Washington, DC, USA, 1980; p. 485. [Google Scholar]
- Lopes, I.F.; Del Lama, M.A.; Del Lama, S.N. Genetic Variability in Three Amazon Parrot Species. Braz. J. Biol. 2007, 67, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol. 2016, 17, 1–11. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, Y.; Ying, C.; Wang, D.; Du, C. Nanopore-Based Fourth-Generation DNA Sequencing Technology. Genom. Proteom. Bioinform. 2015, 13, 4–16. [Google Scholar] [CrossRef]
- Cann, R.L.; Stoneking, M.; Wilson, A.C. Mitochondrial DNA and Human Evolution. Nature 1987, 325, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Boore, J.L.; Brown, W.M. Big Trees from Little Genomes: Mitochondrial Gene Order as a Phylogenetic Tool. Curr. Opin. Genet. Dev. 1998, 6, 668–674. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | DIVALIKE (A) | DIVALIKE + J (B) | DIVALIKE2 (C) | DIVALIKE + J2(D) | DEC (E) | DEC + J (F) | BAYAREALIKE (G) | BAYAREALIKE + J (H) | |
|---|---|---|---|---|---|---|---|---|---|
| Model ID | A | B | C | D | E | F | G | H | |
| Feature | |||||||||
| 1 | Common MJ gene pool | + | + | + | + | + | − | + | − |
| 2 | Common MY gene pool | − | − | − | − | − | + | − | + |
| 3 | Sympatric Speciation in J | − | + | + | + | − | − | − | + |
| 4 | Common CJ gene pool | + | − | − | − | + | − | − | − |
| 5 | Speciation after J to C dispersal | − | − | − | − | − | + | − | − |
| 6 | Return dispersal from C to J | − | − | − | − | − | + | − | − |
| 7 | Stepping stone dispersal from C to H to P | + | + | + | + | − | + | − | + |
| 8 | Common CH gene pool | − | − | − | − | + | − | − | − |
| 9 | Common JH gene pool | − | − | − | − | − | − | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolchanova, S.; Komissarov, A.; Kliver, S.; Mazo-Vargas, A.; Afanador, Y.; Velez-Valentín, J.; de la Rosa, R.V.; Castro-Marquez, S.; Rivera-Colon, I.; Majeske, A.J.; et al. Molecular Phylogeny and Evolution of Amazon Parrots in the Greater Antilles. Genes 2021, 12, 608. https://doi.org/10.3390/genes12040608
Kolchanova S, Komissarov A, Kliver S, Mazo-Vargas A, Afanador Y, Velez-Valentín J, de la Rosa RV, Castro-Marquez S, Rivera-Colon I, Majeske AJ, et al. Molecular Phylogeny and Evolution of Amazon Parrots in the Greater Antilles. Genes. 2021; 12(4):608. https://doi.org/10.3390/genes12040608
Chicago/Turabian StyleKolchanova, Sofiia, Alexey Komissarov, Sergei Kliver, Anyi Mazo-Vargas, Yashira Afanador, Jafet Velez-Valentín, Ricardo Valentín de la Rosa, Stephanie Castro-Marquez, Israel Rivera-Colon, Audrey J. Majeske, and et al. 2021. "Molecular Phylogeny and Evolution of Amazon Parrots in the Greater Antilles" Genes 12, no. 4: 608. https://doi.org/10.3390/genes12040608
APA StyleKolchanova, S., Komissarov, A., Kliver, S., Mazo-Vargas, A., Afanador, Y., Velez-Valentín, J., de la Rosa, R. V., Castro-Marquez, S., Rivera-Colon, I., Majeske, A. J., Wolfsberger, W. W., Hains, T., Corvelo, A., Martinez-Cruzado, J.-C., Glenn, T. C., Robinson, O., Koepfli, K.-P., & Oleksyk, T. K. (2021). Molecular Phylogeny and Evolution of Amazon Parrots in the Greater Antilles. Genes, 12(4), 608. https://doi.org/10.3390/genes12040608