
Predicting TCR-Epitope Binding Specificity Using Deep Metric Learning and Multimodal Learning

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Positive Binding and Negative Binding Datasets
2.2. CDR3B and Epitope Sequence Representation
2.3. Overview of Model Architecture
2.4. Model Training
2.5. Definition of Classification Tasks and General Model Evaluation Procedure
2.6. Processing of CDR3B Sequences
2.7. Identification of Preprocessing Artifacts as Confounding Factors
2.8. Model Evaluation for Specific Seen Epitope Sets
2.9. Model Interpretation
2.10. TCR-pMHC Structure Analysis
3. Results
3.1. A Hybrid Neural Network Represents CDR3B and Epitope Sequences in a Shared Latent Space
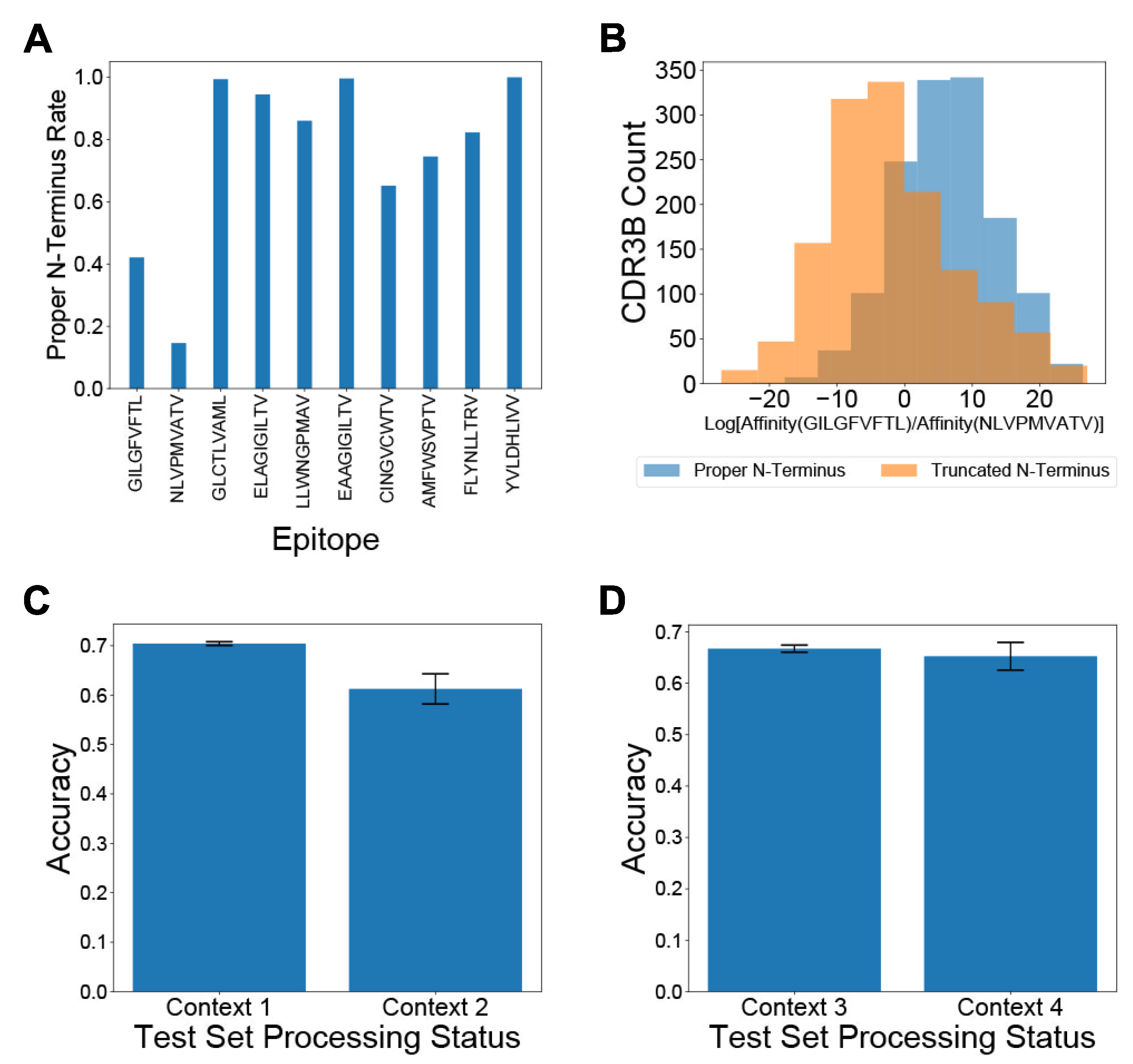
3.2. Artifacts in Preprocessing CDR3B Sequences Confound TCR-Epitope Binding Predictions
3.3. The Trained Model Predicts TCR-Epitope Binding under Task 1 and Task 2 Settings
3.4. Model Interpretation Method Identifies Salient CDR3B Sequence Motifs and Amino Acid Positions Specifying Epitope Binding
3.5. Salient Positions in CDR3B Sequence Do Not Necessarily Reflect Physical Proximity to Epitope
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krogsgaard, M.; Davis, M.M. How T cells ‘see’ antigen. Nat. Immunol. 2005, 6, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Slota, M.; Lim, J.B.; Dang, Y.; Disis, M.L. ELISpot for measuring human immune responses to vaccines. Expert Rev. Vaccines 2011, 10, 299–306. [Google Scholar] [CrossRef]
- Calarota, S.A.; Baldanti, F. Enumeration and characterization of human memory T cells by enzyme-linked immunospot assays. Clin. Dev. Immunol. 2013, 2013, 637649. [Google Scholar] [CrossRef] [PubMed]
- Bacher, P.; Scheffold, A. Flow-cytometric analysis of rare antigen-specific T cells. Cytometry A 2013, 83, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, P.K.; Melenhorst, J.J.; Ladell, K.; Gostick, E.; Scheinberg, P.; Barrett, A.J.; Wooldridge, L.; Roederer, M.; Sewell, A.K.; Price, D.A. Techniques to improve the direct ex vivo detection of low frequency antigen-specific CD8+ T cells with peptide-major histocompatibility complex class I tetramers. Cytometry A 2008, 73, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Kula, T.; Dezfulian, M.H.; Wang, C.I.; Abdelfattah, N.S.; Hartman, Z.C.; Wucherpfennig, K.W.; Lyerly, H.K.; Elledge, S.J. T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 2019, 178, 1016–1028.e1013. [Google Scholar] [CrossRef] [PubMed]
- Benichou, J.; Ben-Hamo, R.; Louzoun, Y.; Efroni, S. Rep-Seq: Uncovering the immunological repertoire through next-generation sequencing. Immunology 2012, 135, 183–191. [Google Scholar] [CrossRef]
- De Simone, M.; Rossetti, G.; Pagani, M. Single Cell T Cell Receptor Sequencing: Techniques and Future Challenges. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef]
- Karosiene, E.; Lundegaard, C.; Lund, O.; Nielsen, M. NetMHCcons: A consensus method for the major histocompatibility complex class I predictions. Immunogenetics 2012, 64, 177–186. [Google Scholar] [CrossRef]
- Nielsen, M.; Andreatta, M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016, 8, 33. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Bennink, J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999, 17, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Glanville, J.; Huang, H.; Nau, A.; Hatton, O.; Wagar, L.E.; Rubelt, F.; Ji, X.; Han, A.; Krams, S.M.; Pettus, C.; et al. Identifying specificity groups in the T cell receptor repertoire. Nature 2017, 547, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.; Fiore-Gartland, A.J.; Hertz, T.; Wang, G.C.; Sharma, S.; Souquette, A.; Crawford, J.C.; Clemens, E.B.; Nguyen, T.H.O.; Kedzierska, K.; et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 2017, 547, 89–93. [Google Scholar] [CrossRef]
- Lanzarotti, E.; Marcatili, P.; Nielsen, M. Identification of the cognate peptide-MHC target of T cell receptors using molecular modeling and force field scoring. Mol. Immunol. 2018, 94, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.P.; Singh, N.K.; Pierce, B.G.; Weng, Z.; Baker, B.M. Computational Modeling of T Cell Receptor Complexes. Methods Mol. Biol. 2016, 1414, 319–340. [Google Scholar] [CrossRef]
- Pierce, B.G.; Weng, Z. A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci. 2013, 22, 35–46. [Google Scholar] [CrossRef]
- Roomp, K.; Domingues, F.S. Predicting interactions between T cell receptors and MHC-peptide complexes. Mol. Immunol. 2011, 48, 553–562. [Google Scholar] [CrossRef]
- Jokinen, E.; Huuhtanen, J.; Mustjoki, S.; Heinonen, M.; Lähdesmäki, H. Determining epitope specificity of T cell receptors with TCRGP. bioRxiv 2019, 542332. [Google Scholar] [CrossRef]
- Jurtz, V.I.; Jessen, L.E.; Bentzen, A.K.; Jespersen, M.C.; Mahajan, S.; Vita, R.; Jensen, K.K.; Marcatili, P.; Hadrup, S.R.; Peters, B.; et al. NetTCR: Sequence-based prediction of TCR binding to peptide-MHC complexes using convolutional neural networks. bioRxiv 2018, 433706. [Google Scholar] [CrossRef]
- Moris, P.; De Pauw, J.; Postovskaya, A.; Ogunjimi, B.; Laukens, K.; Meysman, P. Treating biomolecular interaction as an image classification problem—A case study on T-cell receptor-epitope recognition prediction. bioRxiv 2019. [Google Scholar] [CrossRef]
- Springer, I.; Besser, H.; Tickotsky-Moskovitz, N.; Dvorkin, S.; Louzoun, Y. Prediction of Specific TCR-Peptide Binding From Large Dictionaries of TCR-Peptide Pairs. Front. Immunol. 2020, 11, 1803. [Google Scholar] [CrossRef] [PubMed]
- Petrova, G.; Ferrante, A.; Gorski, J. Cross-reactivity of T cells and its role in the immune system. Crit. Rev. Immunol. 2012, 32, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, Y.; Leung, T.; Rosenberg, C.; Wang, J.; Philbin, J.; Chen, B.; Wu, Y. Learning Fine-Grained Image Similarity with Deep Ranking. In Proceedings of the 2014 IEEE Conference on Computer Vision and Pattern Recognition, Columbus, OH, USA, 23–28 June 2014; pp. 1386–1393. [Google Scholar]
- Hoffer, E.; Ailon, N. Deep Metric Learning Using Triplet Network. In Similarity-Based Pattern Recognition. SIMBAD 2015. Lecture Notes in Computer Science; Springer: Cham, Switzerland, 2014; Volume 9370. [Google Scholar] [CrossRef]
- Kaya, M.; Bilge, H. Deep Metric Learning: A Survey. Symmetry 2019, 11, 1066. [Google Scholar] [CrossRef]
- Baltrusaitis, T.; Ahuja, C.; Morency, L.-P. Multimodal Machine Learning: A Survey and Taxonomy. IEEE Trans. Pattern Anal. Mach. Intell. 2019, 41, 423–443. [Google Scholar] [CrossRef]
- Atchley, W.R.; Zhao, J.; Fernandes, A.D.; Drüke, T. Solving the protein sequence metric problem. Proc. Natl. Acad. Sci. USA 2005, 102, 6395–6400. [Google Scholar] [CrossRef]
- Bagaev, D.V.; Vroomans, R.M.A.; Samir, J.; Stervbo, U.; Rius, C.; Dolton, G.; Greenshields-Watson, A.; Attaf, M.; Egorov, E.S.; Zvyagin, I.V.; et al. VDJdb in 2019: Database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res. 2020, 48, D1057–D1062. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune epitope database—analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef]
- Tickotsky, N.; Sagiv, T.; Prilusky, J.; Shifrut, E.; Friedman, N. McPAS-TCR: A manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 2017, 33, 2924–2929. [Google Scholar] [CrossRef]
- Finnegan, A.; Song, J.S. Maximum entropy methods for extracting the learned features of deep neural networks. PLoS Comput. Biol. 2017, 13, e1005836. [Google Scholar] [CrossRef]
- Finnegan, A.I.; Kim, S.; Jin, H.; Gapinske, M.; Woods, W.S.; Perez-Pinera, P.; Song, J.S. Epigenetic engineering of yeast reveals dynamic molecular adaptation to methylation stress and genetic modulators of specific DNMT3 family members. Nucleic Acids Res. 2020, 48, 4081–4099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, L.; Liu, K.; Wei, X.; Yang, K.; Du, W.; Wang, S.; Guo, N.; Ma, C.; Luo, L.; et al. PIRD: Pan Immune Repertoire Database. Bioinformatics 2019, 36, 897–903. [Google Scholar] [CrossRef]
- Dean, J.; Emerson, R.O.; Vignali, M.; Sherwood, A.M.; Rieder, M.J.; Carlson, C.S.; Robins, H.S. Annotation of pseudogenic gene segments by massively parallel sequencing of rearranged lymphocyte receptor loci. Genome Med. 2015, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Leem, J.; de Oliveira, S.H.P.; Krawczyk, K.; Deane, C.M. STCRDab: The structural T-cell receptor database. Nucleic Acids Res. 2018, 46, D406–D412. [Google Scholar] [CrossRef]
- Miao, Z.; Cao, Y. Quantifying side-chain conformational variations in protein structure. Sci. Rep. 2016, 6, 37024. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luu, A.M.; Leistico, J.R.; Miller, T.; Kim, S.; Song, J.S. Predicting TCR-Epitope Binding Specificity Using Deep Metric Learning and Multimodal Learning. Genes 2021, 12, 572. https://doi.org/10.3390/genes12040572
Luu AM, Leistico JR, Miller T, Kim S, Song JS. Predicting TCR-Epitope Binding Specificity Using Deep Metric Learning and Multimodal Learning. Genes. 2021; 12(4):572. https://doi.org/10.3390/genes12040572
Chicago/Turabian StyleLuu, Alan M., Jacob R. Leistico, Tim Miller, Somang Kim, and Jun S. Song. 2021. "Predicting TCR-Epitope Binding Specificity Using Deep Metric Learning and Multimodal Learning" Genes 12, no. 4: 572. https://doi.org/10.3390/genes12040572
APA StyleLuu, A. M., Leistico, J. R., Miller, T., Kim, S., & Song, J. S. (2021). Predicting TCR-Epitope Binding Specificity Using Deep Metric Learning and Multimodal Learning. Genes, 12(4), 572. https://doi.org/10.3390/genes12040572