
Deciphering the Monilinia fructicola Genome to Discover Effector Genes Possibly Involved in Virulence


Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. DNA and RNA Isolation
2.3. Sequencing and de Novo Assembly of the Genome
2.4. Genome Annotation and Expression Quantification
2.5. Secondary Metabolite Gene Cluster Analysis
2.6. Secretome and Effector Prediction
2.7. Characterization of Candidate Effector Proteins
2.7.1. Amplification of Candidate Effector Genes
2.7.2. Ligation of Gene Constructs
2.7.3. Transient Effector Gene Expression in Nicotiana benthamiana
3. Results
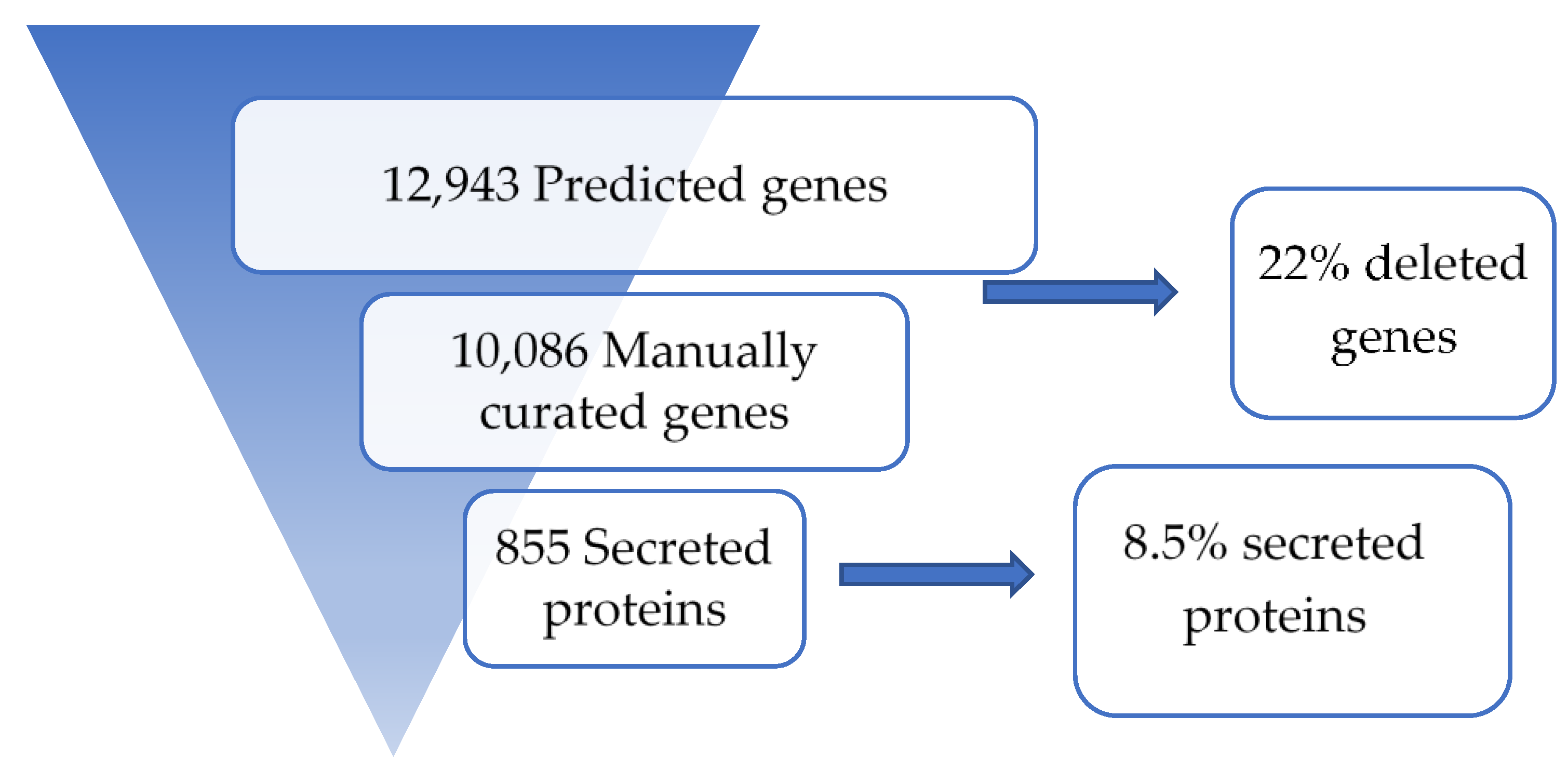
3.1. Sequence Assembly and Annotation
3.2. Secondary Metabolites
3.3. Secreted Proteins
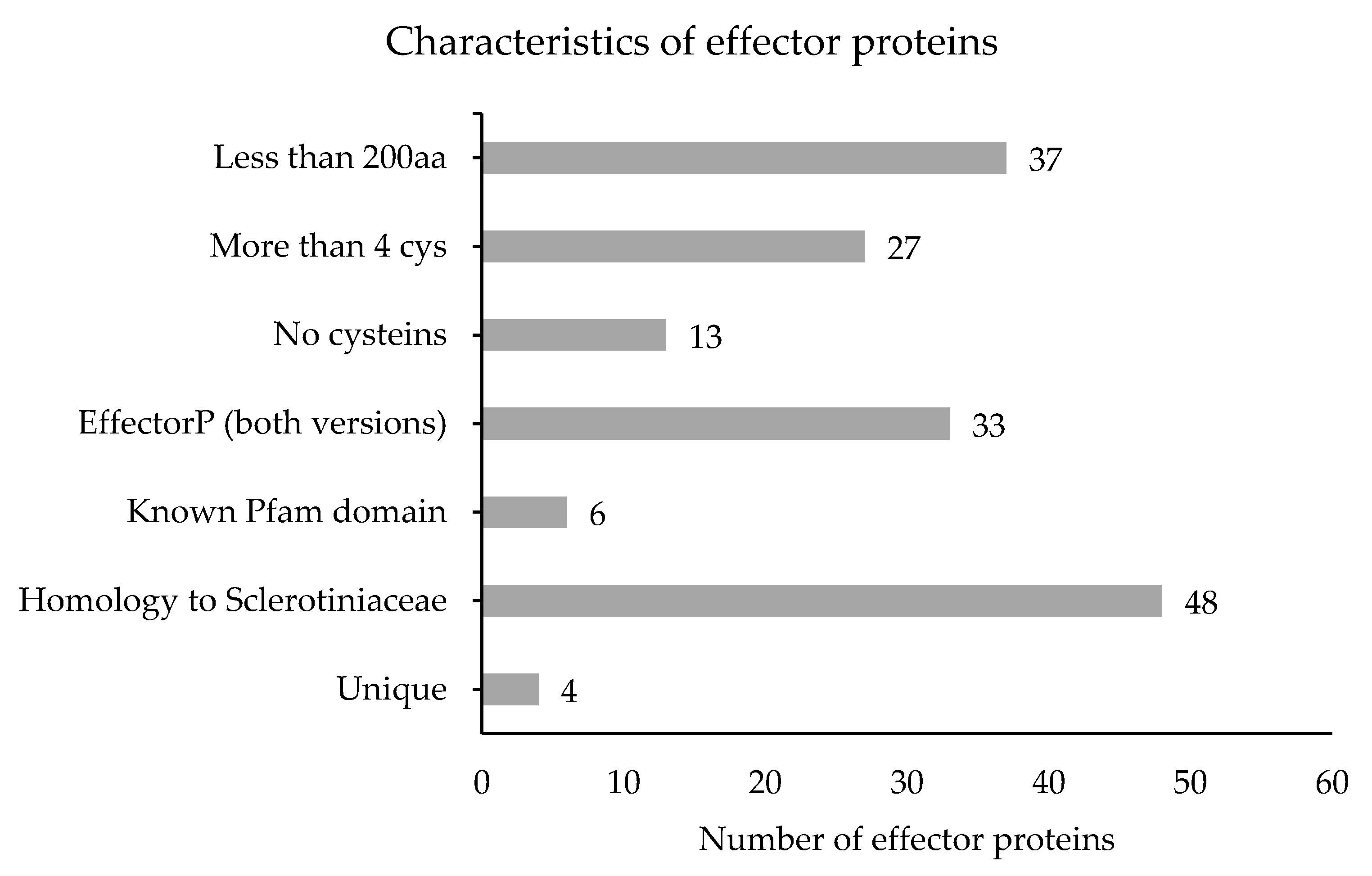
3.4. Effector Proteins
3.5. Characterization of M. fructicola Candidate Effectors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martini, C.; Mari, M. Chapter 7—Monilinia fructicola, Monilinia laxa (Monilinia rot, Brown rot). In Postharvest Decay; Bautista-Baños, S., Ed.; Academic Press: San Diego, CA, USA, 2014; pp. 233–265. [Google Scholar]
- EPPO. Monilinia Fructicola; Bulletin 33; 2003; pp. 281–288. Available online: https://doi.org/10.1046/j.1365-2338.2003.00639.x (accessed on 1 April 2021).
- De Cal, A.; Gell, I.; Usall, J.; Viñas, I.; Melgarejo, P. First report of brown rot caused by Monilinia fructicola in peach orchards in Ebro Valley, Spain. Plant Dis. 2009, 93, 763. [Google Scholar] [CrossRef] [PubMed]
- Villarino, M.; Egüen, B.; Lamarca, N.; Segarra, J.; Usall, J.; Melgarejo, P.; De Cal, A. Occurrence of Monilinia laxa and M. fructigena after introduction of M. fructicola in peach orchards in Spain. Eur. J. Plant Pathol. 2013, 137, 835–845. [Google Scholar] [CrossRef]
- Gotor-Vila, A.; Teixidó, N.; Casals, C.; Torres, R.; De Cal, A.; Guijarro, B.; Usall, J. Biological control of brown rot in stone fruit using Bacillus amyloliquefaciens CPA-8 under field conditions. Crop. Prot. 2017, 102, 72–80. [Google Scholar] [CrossRef]
- Sisquella, M.; Viñas, I.; Picouet, P.; Torres, R.; Usall, J. Effect of host and Monilinia spp. variables on the efficacy of radio frequency treatment on peaches. Postharvest Biol. Technol. 2014, 87, 6–12. [Google Scholar] [CrossRef]
- Usall, J.; Casals, C.; Sisquella, M.; Palou, L.; De Cal, A. Alternative technologies to control postharvest diseases of stone fruits. Stewart Postharvest Rev. 2015, 4, 1–6. [Google Scholar]
- van Kan, J.A. Licensed to kill: The lifestyle of a necrotrophic plant pathogen. Trends Plant Sci. 2006, 11, 247–253. [Google Scholar] [CrossRef]
- Oliver, R.P.; Solomon, P.S. New developments in pathogenicity and virulence of necrotrophs. Curr. Opin. Plant Biol. 2010, 13, 415–419. [Google Scholar] [CrossRef]
- Veloso, J.; van Kan, J.A. Many shades of grey in Botrytis-host plant interactions. Trends Plant Sci. 2018, 23, 613–622. [Google Scholar] [CrossRef]
- Faris, J.D.; Zhang, Z.; Lu, H.; Lu, S.; Reddy, L.; Cloutier, S.; Fellers, J.P.; Meinhardt, S.W.; Rasmussen, J.B.; Xu, S.S.; et al. A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proc. Natl. Acad. Sci. USA 2010, 107, 13544–13549. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Z.; Faris, J.D.; Oliver, R.P.; Syme, R.; McDonald, M.C.; McDonald, B.A.; Solomon, P.S.; Lu, S.; Shelver, W.L.; et al. The cysteine rich necrotrophic effector SnTox1 produced by Stagonospora nodorum triggers susceptibility of wheat lines harboring Snn1. PLoS Pathog. 2012, 8, e1002467. [Google Scholar] [CrossRef]
- Shi, G.; Zhang, Z.; Friesen, T.L.; Raats, D.; Fahima, T.; Brueggeman, R.S.; Lu, S.; Trick, H.N.; Liu, Z.; Chao, W.; et al. The hijacking of a receptor kinase–driven pathway by a wheat fungal pathogen leads to disease. Sci. Adv. 2016, 2, e1600822. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.-C.; Oliver, R.P.; Solomon, P.S.; Moffat, C.S. Proteinaceous necrotrophic effectors in fungal virulence. Funct. Plant Biol. 2010, 37, 907–912. [Google Scholar] [CrossRef]
- Tan, K.-C.; Phan, H.T.T.; Rybak, K.; John, E.; Chooi, Y.-H.; Solomon, P.S.; Oliver, R.P. Functional redundancy of necrotrophic effectors—consequences for exploitation for breeding. Front. Plant Sci. 2015, 6, 501. [Google Scholar] [CrossRef]
- Arenas, Y.C.; Kalkman, E.R.; Schouten, A.; Dieho, M.; Vredenbregt, P.; Uwumukiza, B.; Ruiz, M.O.; van Kan, J.A. Functional analysis and mode of action of phytotoxic Nep1-like proteins of Botrytis cinerea. Physiol. Mol. Plant Pathol. 2010, 74, 376–386. [Google Scholar] [CrossRef]
- Dalmais, B.; Schumacher, J.; Moraga, J.; Le Pêcheur, P.; Tudzynski, B.; Collado, I.G.; Viaud, M. The Botrytis cinerea phytotoxin botcinic acid requires two polyketide synthases for production and has a redundant role in virulence with botrydial. Mol. Plant Pathol. 2011, 12, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Frías, M.; González, C.; Brito, N. BcSpl1, a cerato-platanin family protein, contributes to Botrytis cinerea virulence and elicits the hypersensitive response in the host. New Phytol. 2011, 192, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Benitez, C.; Melgarejo, P.; Sandin-España, P.; Sevilla-Morán, B.; De Cal, A. Degrading enzymes and phytotoxins in Monilinia spp. Eur. J. Plant Pathol. 2019, 154, 305–318. [Google Scholar] [CrossRef]
- Lee, M.-H.; Chiu, C.-M.; Roubtsova, T.; Chou, C.-M.; Bostock, R.M. Overexpression of a redox-regulated cutinase gene, MfCUT1, increases virulence of the brown rot pathogen Monilinia fructicola on Prunus spp. Mol. Plant-Microbe Interact. 2010, 23, 176–186. [Google Scholar] [CrossRef]
- Möller, M.; Stukenbrock, E.H. Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Genet. 2017, 15, 756–771. [Google Scholar] [CrossRef]
- Angelini, R.M.D.M.; Romanazzi, G.; Pollastro, S.; Rotolo, C.; Faretra, F.; Landi, L. New high-quality draft genome of the brown rot fungal pathogen Monilinia fructicola. Genome Biol. Evol. 2019, 11, 2850–2855. [Google Scholar] [CrossRef]
- Rivera, Y.; Zeller, K.; Srivastava, S.; Sutherland, J.; Galvez, M.; Nakhla, M.; Poniatowska, A.; Schnabel, G.; Sundin, G.; Abad, Z.G. Draft genome resources for the phytopathogenic fungi Monilinia fructicola, M. fructigena, M. polystroma, and M. laxa, the causal agents of brown rot. Phytopathology 2018, 108, 1141–1142. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, L.; Abuduaini, A.; Zhou, H.; Wang, Y.; Suo, F. Complete mitochondrial genome of plant pathogen Monilinia fructicola (Sclerotiniaceae, Helotiales). Mitochondrial DNA Part B 2019, 4, 791–792. [Google Scholar] [CrossRef]
- Vilanova, L.; Wisniewski, M.; Norelli, J.; Viñas, I.; Torres, R.; Usall, J.; Phillips, J.; Droby, S.; Teixidó, N. Transcriptomic profiling of apple in response to inoculation with a pathogen (Penicillium expansum) and a non-pathogen (Penicillium digitatum). Plant Mol. Biol. Rep. 2013, 32, 566–583. [Google Scholar] [CrossRef]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Baldwin-Brown, J.G.; Long, A.D.; Emerson, J.J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016, 44, e147. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Min, B.; Grigoriev, I.V.; Choi, I.-G. FunGAP: Fungal Genome Annotation Pipeline using evidence-based gene model evaluation. Bioinformatics 2017, 33, 2936–2937. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2007, 18, 188–196. [Google Scholar] [CrossRef]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS: Table 1. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef]
- Pedro, H.; Yates, A.D.; Kersey, P.J.; De Silva, N.H. Collaborative annotation redefines gene sets for crucial phytopathogens. Front. Microbiol. 2019, 10, 2477. [Google Scholar] [CrossRef]
- Van Kan, J.A.L.; Stassen, J.H.M.; Mosbach, A.; Van Der Lee, T.A.J.; Faino, L.; Farmer, A.D.; Papasotiriou, D.G.; Zhou, S.; Seidl, M.F.; Cottam, E.; et al. A gapless genome sequence of the fungus Botrytis cinerea. Mol. Plant Pathol. 2016, 18, 75–89. [Google Scholar] [CrossRef]
- Palmer, J.; Stajich, J. Nextgenusfs/Funannotate: Funannotate v1.5.3 (Version 1.5.3). Zenodo 2019. Available online: https://zenodo.org/record/2604804#.YHAWwj9cJPZ (accessed on 15 June 2018).
- Valero-Jiménez, C.A.; Steentjes, M.B.F.; Slot, J.C.; Shi-Kunne, X.; Scholten, O.E.; Kan, J.A.L.V. Dynamics in Secondary Metabolite Gene Clusters in Otherwise Highly Syntenic and Stable Genomes in the Fungal Genus Botrytis. Genome Biol. Evol. 2020, 12, 2491–2507. [Google Scholar] [CrossRef]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. antiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; Santos, E.L.C.D.L.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; De Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, O.; Brunak, S.; Von Heijne, G.; Nielsen, H.A. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Dodds, P.N.; Gardiner, D.M.; Singh, K.B.; Taylor, J.M. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol. Plant Pathol. 2018, 19, 2094–2110. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Gardiner, D.M.; Dodds, P.N.; Tini, F.; Covarelli, L.; Singh, K.B.; Manners, J.M.; Taylor, J.M. EffectorP: Predicting fungal effector proteins from secretomes using machine learning. New Phytol. 2016, 210, 743–761. [Google Scholar] [CrossRef] [PubMed]
- Kohn, L. Monilinia Fructicola Strain LMK 125 Scaffold_001, Whole Genome Shotgun Sequence. GenBank: NGKE01000001.1. 2017. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NGKE01000001 (accessed on 15 September 2020).
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Genet. 2019, 17, 167–180. [Google Scholar] [CrossRef] [PubMed]
- De Cal, A.; Melgarejo, P. Effects of pyroquilon on the infection process of Monilinia laxa causing peach twig blight. Pestic. Sci. 1993, 39, 267–269. [Google Scholar] [CrossRef]
- Schumacher, J. DHN melanin biosynthesis in the plant pathogenic fungus Botrytis cinerea is based on two developmentally regulated key enzyme (PKS)-encoding genes. Mol. Microbiol. 2015, 99, 729–748. [Google Scholar] [CrossRef]
- Schouten, A.; Van Baarlen, P.; Van Kan, J.A.L. Phytotoxic Nep1-like proteins from the necrotrophic fungus Botrytis cinerea associate with membranes and the nucleus of plant cells. New Phytol. 2007, 177, 493–505. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Rodríguez-Píres, S.; Torres, R.; De Cal, A.; Usall, J.; Gabaldón, T. Genome esquence of the brown rot fungal pathogen Monilinia laxa. Genome Announc. 2018, 6, e00214-18. [Google Scholar] [CrossRef]
- Landi, L.; Angelini, R.M.D.M.; Pollastro, S.; Abate, D.; Faretra, F.; Romanazzi, G. Genome sequence of the brown rot fungal pathogen Monilinia fructigena. BMC Res. Notes 2018, 11, 1–3. [Google Scholar] [CrossRef]
- Derbyshire, M.; Denton-Giles, M.; Hegedus, D.; Seifbarghy, S.; Rollins, J.; Van Kan, J.; Seidl, M.F.; Faino, L.; Mbengue, M.; Navaud, O.; et al. The complete genome sequence of the phytopathogenic fungus Sclerotinia sclerotiorum reveals insights into the genome architecture of broad host range pathogens. Genome Biol. Evol. 2017, 9, 593–618. [Google Scholar] [CrossRef]
- Amselem, J.; Cuomo, C.A.; Van Kan, J.A.L.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; De Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef]
- Marcet-Houben, M.; Ballester, A.-R.; De La Fuente, B.; Harries, E.; Marcos, J.F.; González-Candelas, L.; Gabaldón, T. Genome sequence of the necrotrophic fungus Penicillium digitatum, the main postharvest pathogen of citrus. BMC Genom. 2012, 13, 646. [Google Scholar] [CrossRef]
- Baró-Montel, N.; Vall-Llaura, N.; Usall, J.; Teixidó, N.; Naranjo-Ortíz, M.A.; Gabaldón, T.; Torres, R. Pectin methyl esterases and rhamnogalacturonan hydrolases: Weapons for successful Monilinia laxa infection in stone fruit? Plant Pathol. 2019, 68, 1381–1393. [Google Scholar] [CrossRef]
- Rodríguez-Pires, S.; Melgarejo, P.; De Cal, A.; Espeso, E.A. Pectin as carbon source for Monilinia laxa exoproteome and expression profiles of related genes. Mol. Plant-Microbe Interact. 2020, 33, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Petrasch, S.; Silva, C.J.; Mesquida-Pesci, S.D.; Gallegos, K.; Abeele, C.V.D.; Papin, V.; Fernandez-Acero, F.J.; Knapp, S.J.; Blanco-Ulate, B. Infection strategies deployed by Botrytis cinerea, Fusarium acuminatum, and Rhizopus stolonifer as a function of tomato fruit ripening stage. Front. Plant Sci. 2019, 10, 223. [Google Scholar] [CrossRef]
- Valero-Jiménez, C.A.; Veloso, J.; Staats, M.; Van Kan, J.A.L. Comparative genomics of plant pathogenic Botrytis species with distinct host specificity. BMC Genom. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Rodríguez-Pires, S.; Melgarejo, P.; De Cal, A.; Espeso, E.A. Proteomic studies to understand the mechanisms of peach tissue degradation by Monilinia laxa. Front. Plant Sci. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Balsells-Llauradó, M.; Silva, C.J.; Usall, J.; Vall-Llaura, N.; Serrano-Prieto, S.; Teixidó, N.; Mesquida-Pesci, S.D.; De Cal, A.; Blanco-Ulate, B.; Torres, R. Depicting the battle between nectarine and Monilinia laxa: The fruit developmental stage dictates the effectiveness of the host defenses and the pathogen’s infection strategies. Hortic. Res. 2020, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kars, I.; McCalman, M.; Wagemakers, L.; Van Kan, J.A.L. Functional analysis of Botrytis cinerea pectin methylesterase genes by PCR-based targeted mutagenesis: Bcpme1 and Bcpme2 are dispensable for virulence of strain B05.10. Mol. Plant Pathol. 2005, 6, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Have, A.T.; Mulder, W.; Visser, J.; Van Kan, J.A.L. The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea. Mol. Plant-Microbe Interact. 1998, 11, 1009–1016. [Google Scholar] [CrossRef]
- Staats, M.; van Baarlen, P.; Schouten, A.; van Kan, J.A.L. Functional analysis of NLP genes from Botrytis elliptica. Mol. Plant Pathol. 2007, 8, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lin-Wang, K.; Wang, H.; Gu, C.; Dare, A.P.; Espley, R.V.; He, H.; Allan, A.C.; Han, Y. Molecular genetics of blood-fleshed peach reveals activation of anthocyanin biosynthesis by NAC transcription factors. Plant J. 2015, 82, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Leisen, T.; Bietz, F.; Werner, J.; Wegner, A.; Schaffrath, U.; Scheuring, D.; Willmund, F.; Mosbach, A.; Scalliet, G.; Hahn, M. CRISPR/Cas with ribonucleoprotein complexes and transiently selected telomere vectors allows highly efficient marker-free and multiple genome editing in Botrytis cinerea. PLoS Pathog. 2020, 16, e1008326. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Genome Size (Mb) | Number of Contigs | Contig N50 (Kb) | Contig L50 (bp) | GC (%) Fraction | BUSCO Complete (Partial) | Predicted Genes | Reference |
|---|---|---|---|---|---|---|---|---|
| CMPC6 | 42.95 | 98 | 988 | 14 | 41.5 | 98.7 (99.5) | 10,086 | This work |
| Mfrc123 | 44.05 | 20 | 2592 | 7 | 40.8 | 88 (98) | 13,749 | De Miccolis Angelini et al. (2019) |
| BR-32 | 42.82 | 2643 | 62 | 205 | 41.7 | >97% | NA 1 | Rivera et al. (2018) |
| LMK 125 | 44.68 | 3535 | 37 | 353 | 40.1 | NA 1 | NA 1 | Kohn (2017) |
| Fungal Species | PCWDE | FPCWDE | References | |||||
|---|---|---|---|---|---|---|---|---|
| Total | C | H | HP | P | H/HP/P | |||
| M. fructicola | 90 | 18 | 27 | 12 | 33 | 72 | 22 | This work |
| S. sclerotiorum | 106 | 20 | 40 | 13 | 33 | 86 | 32 | Anselem et al., 2010 |
| B. aclada | 89 | 17 | 27 | 13 | 32 | 72 | 31 | Valero-Jiménez et al., 2020 |
| B. cinerea T4 | 118 | 18 | 41 | 15 | 44 | 100 | 36 | Anselem et al., 2010 |
| Neurospora crassa | 78 | 26 | 31 | 13 | 8 | 52 | 22 | Anselem et al., 2010 |
| Penicillium digitatum | 49 | NA 1 | NA 1 | NA 1 | NA 1 | NA 1 | NA 1 | Marcet-Houben et al., 2012 |
| Effector | Protein Size (aa) | # Cys | In Planta CPM | In Vitro CPM | Ratio P/V | PFAM Domain | Homology to Other Species |
|---|---|---|---|---|---|---|---|
| MFRU_001g01030 | 232 | 13 | 14.8 | 123 | 0.12 | Botrytis, Sclerotinia | |
| MFRU_001g04320 | 95 | 0 | 197 | 130 | 1.5 | Many fungi | |
| MFRU_001g04590 | 122 | 6 | 34.3 | 84 | 0.4 | Many fungi | |
| MFRU_001g05460 | 148 | 8 | 16.7 | 154 | 0.11 | Botrytis, Sclerotinia, Chaetomium, Rutstroemia | |
| MFRU_002g00070 | 246 | 0 | 9.8 | 0.6 | 15.2 | Botrytis | |
| MFRU_002g02190 | 94 | 10 | 150 | 0.8 | 181 | Aspergillus, Botrytis, Sclerotinia | |
| MFRU_002g03250 | 203 | 0 | 50 | 76 | 0.7 | Many fungi | |
| MFRU_002g05260 1 | 221 | 10 | 7.3 | 8.0 | 0.9 | PF05730 (CFEM domain) | Botrytis, Sclerotinia |
| MFRU_003g02920 | 190 | 6 | 2.1 | 3.1 | 0.7 | Periconia, Sclerotinia | |
| MFRU_003g05140 | 129 | 2 | 2.4 | 16.8 | 0.14 | Botrytis, Sclerotinia | |
| MFRU_004g01490 | 109 | 2 | 11.7 | 45 | 0.3 | None | |
| MFRU_004g01570 | 180 | 4 | 1.4 | 1.3 | 1.0 | None | |
| MFRU_004g02710 | 105 | 6 | 2.8 | 1.3 | 2.1 | Aspergillus, Cladophora, Sclerotinia | |
| MFRU_004g02780 | 87 | 0 | 0.9 | 0.6 | 1.4 | None | |
| MFRU_005g03220 | 83 | 7 | 1 | 0.9 | 1.1 | Botrytis, Sclerotinia | |
| MFRU_008g03050 | 113 | 7 | 3.1 | 611 | 0.01 | Botrytis, Sclerotinia | |
| MFRU_008g03430 | 204 | 0 | 3.3 | 8.9 | 0.4 | Pochonia, Purpureocillium, Sclerotinia | |
| MFRU_009g00790 | 177 | 2 | 0.7 | 0.8 | 1.0 | Many fungi | |
| MFRU_012g00980 | 173 | 6 | 4.8 | 263 | 0.02 | Many fungi | |
| MFRU_012g01440 1 | 245 | 4 | 25.3 | 325 | 0.08 | PF14021 (tuberculosis necrotizing toxin) | Many fungi |
| MFRU_012g01930 | 191 | 4 | 59 | 8.7 | 6.7 | Botrytis, Rutstroemia, Sclerotinia | |
| MFRU_014g01140 | 160 | 8 | 1 | 0.7 | 1.4 | Botrytis, Sclerotinia | |
| MFRU_014g02060 | 247 | 3 | 4.1 | 9.5 | 0.4 | PF05630 (necrosis-inducing protein NPP1) | Many fungi |
| MFRU_015g00570 | 92 | 8 | 5.4 | 200 | 0.03 | Many fungi | |
| MFRU_017g01630 | 128 | 7 | 15.9 | 138 | 0.1 | Botrytis, Diplocarpon, Rutstroemia, Sclerotinia | |
| MFRU_022g00070 | 192 | 9 | 1.9 | 0.8 | 2.2 | Botrytis | |
| MFRU_028g01250 | 149 | 5 | 3.6 | 35.4 | 0.12 | PF07249 (cerato-platanin) | Many fungi |
| MFRU_030g00580 1 | 160 | 6 | 135 | 14.9 | 9.1 | Botrytis, Monilinia | |
| MFRU_034g00500 1 | 82 | 8 | 201 | 52.7 | 3.8 | Botrytis | |
| MFRU_035g00290 | 167 | 8 | 10.9 | 8.7 | 1.2 | Botrytis, Sclerotinia | |
| MFRU_036g00390 | 263 | 18 | 6.7 | 49.7 | 0.13 | None | |
| MFRU_048g00370 1 | 147 | 9 | 4.4 | 1.7 | 2.6 | Many fungi | |
| MFRU_062g00230 | 126 | 0 | 1.0 | 0.8 | 1.2 | Botrytis, Monilinia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilanova, L.; Valero-Jiménez, C.A.; van Kan, J.A.L. Deciphering the Monilinia fructicola Genome to Discover Effector Genes Possibly Involved in Virulence. Genes 2021, 12, 568. https://doi.org/10.3390/genes12040568
Vilanova L, Valero-Jiménez CA, van Kan JAL. Deciphering the Monilinia fructicola Genome to Discover Effector Genes Possibly Involved in Virulence. Genes. 2021; 12(4):568. https://doi.org/10.3390/genes12040568
Chicago/Turabian StyleVilanova, Laura, Claudio A. Valero-Jiménez, and Jan A.L. van Kan. 2021. "Deciphering the Monilinia fructicola Genome to Discover Effector Genes Possibly Involved in Virulence" Genes 12, no. 4: 568. https://doi.org/10.3390/genes12040568
APA StyleVilanova, L., Valero-Jiménez, C. A., & van Kan, J. A. L. (2021). Deciphering the Monilinia fructicola Genome to Discover Effector Genes Possibly Involved in Virulence. Genes, 12(4), 568. https://doi.org/10.3390/genes12040568