Intragenic Deletions in FLNB Are Part of the Mutational Spectrum Causing Spondylocarpotarsal Synostosis Syndrome

 , ,
, , {kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects and Ethical Consent
2.2. Sequence Analysis
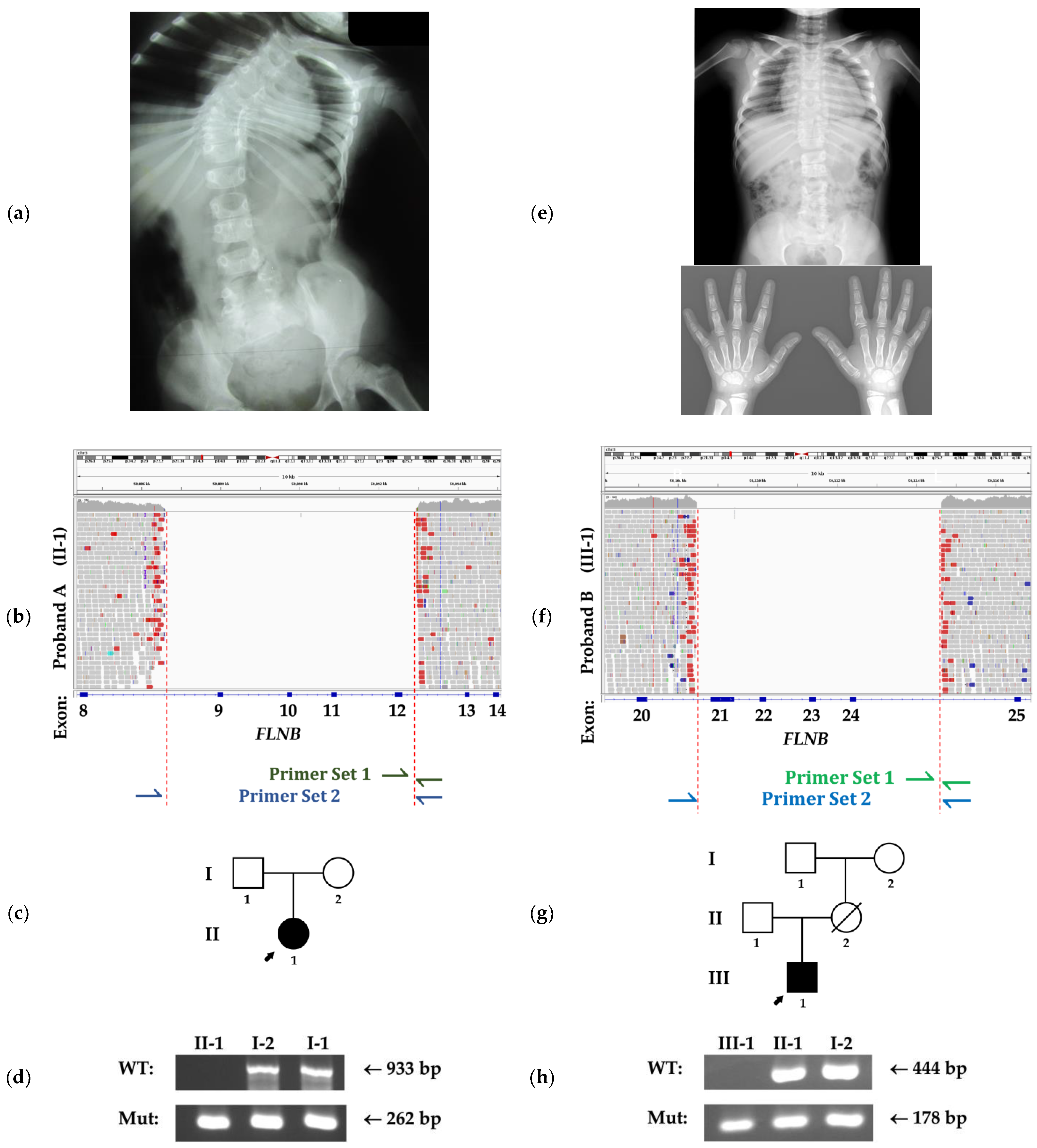
3. Clinical Reports
3.1. Family A
3.2. Family B
4. Results
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krakow, D.; Robertson, S.P.; King, L.M.; Morgan, T.; Sebald, E.T.; Bertolotto, C.; Wachsmann-Hogiu, S.; Acuna, D.; Shapiro, S.S.; Takafuta, T. Mutations in the gene encoding Filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat. Genet. 2004, 36, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Carapito, R.; Goldenberg, A.; Paul, N.; Pichot, A.; David, A.; Hamel, A.; Dumant-Forest, C.; Leroux, J.; Ory, B.; Isidor, B. Protein-altering MYH3 variants are associated with a spectrum of phenotypes extending to Spondylocarpotarsal Synostosis syndrome. Eur. J. Hum. Genet. 2016, 24, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Watanabe, S.; Kinoshita, A.; Mishima, H.; Nishimura, G.; Moriuchi, H.; Yoshiura, K.; Dateki, S. Identification of a homozygous frameshift variant in RFLNA in a patient with a typical phenotype of Spondylocarpotarsal Synostosis Syndrome. J. Hum. Genet. 2019, 64, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Esposito, V.; De Brasi, D.; Mattiacci, D.M.; Krakow, D.; Lee, B.; Salerno, M. Spondylocarpotarsal Synostosis: Long-term follow-up of a case due to FLNB mutations. Am. J. Med. Genet. A 2008, 146, 1230. [Google Scholar] [CrossRef] [PubMed]
- Farrington-Rock, C.; Kirilova, V.; Dillard-Telm, L.; Borowsky, A.D.; Chalk, S.; Rock, M.J.; Cohn, D.H.; Krakow, D. Disruption of the FLNB gene in mice phenocopies the human disease Spondylocarpotarsal Synostosis syndrome. Hum. Mol. Genet. 2008, 17, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Mitter, D.; Krakow, D.; Farrington-Rock, C.; Meinecke, P. Expanded clinical spectrum of Spondylocarpotarsal Synostosis syndrome and possible manifestation in a heterozygous father. Am. J. Med. Genet. A 2008, 146, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, C.; Siong H’ng, W.; Chang, C.; Lin, W.; Chen, Y.; Wu, J.; Tsai, F. Filamin B Loss-of-function mutation in dimerization domain causes autosomal-recessive Spondylocarpotarsal Synostosis Syndrome with rib anomalies. Hum. Mutat. 2017, 38, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Salian, S.; Shukla, A.; Shah, H.; Bhat, S.N.; Bhat, V.R.; Nampoothiri, S.; Shenoy, R.; Phadke, S.; Hariharan, S.; Girisha, K. Seven additional families with Spondylocarpotarsal Synostosis syndrome with novel biallelic deleterious variants in FLNB. Clin. Genet. 2018, 94, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Yasin, S.; Makitie, O.; Naz, S. Spondylocarpotarsal Synostosis syndrome due to a novel loss of function FLNB variant: A case report. BMC Musculoskelet. Disord. 2021, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, L.; Morgan, T.; Bonafe, L.; Wessels, M.; Bialer, M.; Willems, P.; Cohn, D.; Krakow, D.; Robertson, S. Mutations in FLNB cause boomerang dysplasia. J. Med. Genet. 2005, 42, e43. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Parthasarathy, P.; Halliday, B.J.; Morgan, T.; Dean, J.; Nowaczyk, M.J.; Markie, D.; Robertson, S.P.; Wade, E.M. Deletion of exon 1 in AMER1 in osteopathia striata with cranial sclerosis. Genes 2020, 11, 1439. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. BCFtools/RoH: A hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Yengo, L.; Wray, N.R.; Visscher, P.M. Extreme inbreeding in a European ancestry sample from the contemporary UK population. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kotzot, D. Complex and segmental Uniparental Disomy (UPD): Review and lessons from rare chromosomal complements. J. Med. Genet. 2001, 38, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lian, G.; Lenkinski, R.; De Grand, A.; Vaid, R.R.; Bryce, T.; Stasenko, M.; Boskey, A.; Walsh, C.; Sheen, V. Filamin B mutations cause chondrocyte defects in skeletal development. Hum. Mol. Genet. 2007, 16, 1661–1675. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukushima, K.; Parthasarathy, P.; Wade, E.M.; Morgan, T.; Gowrishankar, K.; Markie, D.M.; Robertson, S.P. Intragenic Deletions in FLNB Are Part of the Mutational Spectrum Causing Spondylocarpotarsal Synostosis Syndrome. Genes 2021, 12, 528. https://doi.org/10.3390/genes12040528
Fukushima K, Parthasarathy P, Wade EM, Morgan T, Gowrishankar K, Markie DM, Robertson SP. Intragenic Deletions in FLNB Are Part of the Mutational Spectrum Causing Spondylocarpotarsal Synostosis Syndrome. Genes. 2021; 12(4):528. https://doi.org/10.3390/genes12040528
Chicago/Turabian StyleFukushima, Kaya, Padmini Parthasarathy, Emma M. Wade, Tim Morgan, Kalpana Gowrishankar, David M. Markie, and Stephen P. Robertson. 2021. "Intragenic Deletions in FLNB Are Part of the Mutational Spectrum Causing Spondylocarpotarsal Synostosis Syndrome" Genes 12, no. 4: 528. https://doi.org/10.3390/genes12040528
APA StyleFukushima, K., Parthasarathy, P., Wade, E. M., Morgan, T., Gowrishankar, K., Markie, D. M., & Robertson, S. P. (2021). Intragenic Deletions in FLNB Are Part of the Mutational Spectrum Causing Spondylocarpotarsal Synostosis Syndrome. Genes, 12(4), 528. https://doi.org/10.3390/genes12040528