
Scale and Scope of Gene-Alcohol Interactions in Chronic Pancreatitis: A Systematic Review

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Disease Definitions
2.2. Research Strategy
2.3. Literature Searches and Selection Processes
2.4. Data Extraction
2.5. Statistical Analysis
2.6. Variant Classification in Accordance with Allele Frequency
3. Results
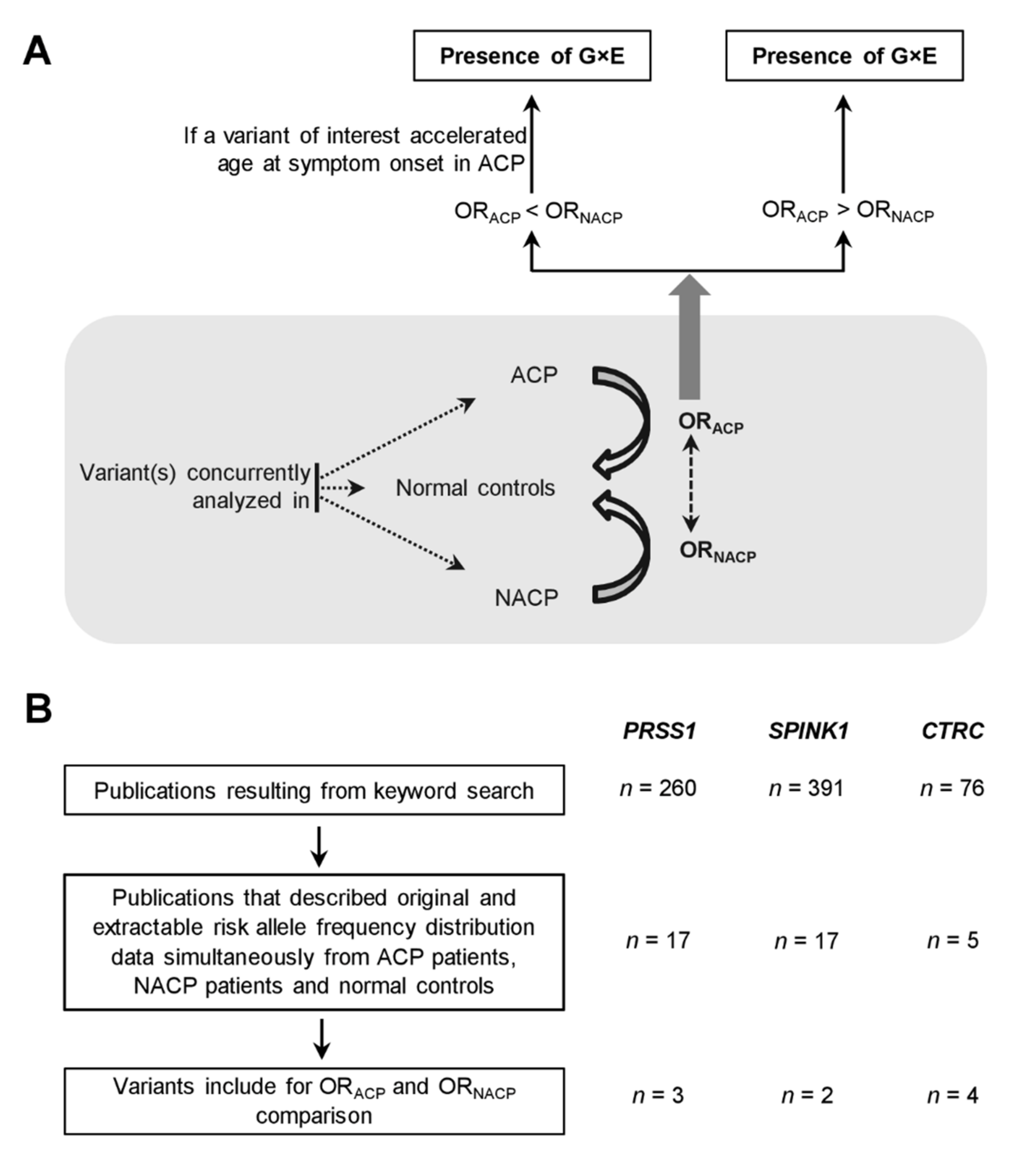
3.1. Variants Included for Analysis
3.2. Evidence Suggesting Extensive G×E Interactions in CP
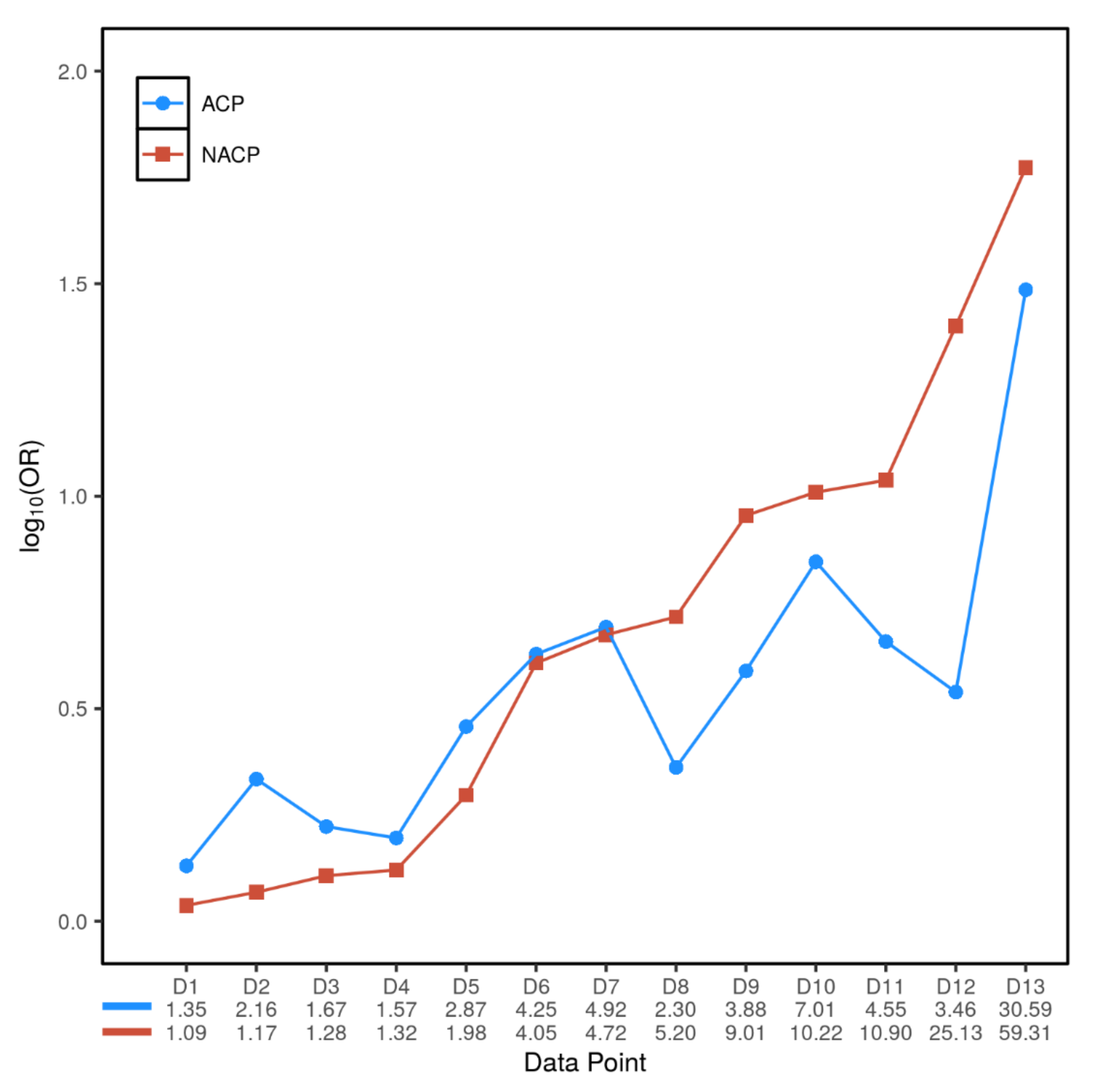
3.3. Inter- and Intra-Variant Comparison of the Paired ORACP and ORNACP Values Revealed a Dichotomized Genetic Effect

{kind=link}
{kind=link}
| Gene or Locus | Variant | Number of ACP/NACP/Normal Controls | Allele Frequency in ACP | Allele Frequency in NACP | Allele Frequency in Normal Controls | ORACP (95% CI); p Value | ORNACP (95% CI); p Value | Source of Data | Data Point Denoted in Figure 2 |
|---|---|---|---|---|---|---|---|---|---|
| CLDN2 | rs12688220 | 42.7% | 32.2% | Not available | Not available | Directly taken from Whitcomb et al. [12] | |||
| rs7057398 | 1.57 (1.14–2.18) | 1.32 (1.15–1.51) | Directly taken from Derikx et al. [40] | D4 | |||||
| CPA1 | Aggregate variants with apparent activity <20% | 456/944/3938 | 0.2% | 1.5% | 0.06% | 3.46 (0.67–17.84); | 25.13 (9.72–65.02); | Witt et al. [11]; only German data were used. | D12 |
| (2/912) | (29/1888) | (5/7878) | p = 0.34 | p < 2.2 × 10−16 | |||||
| CEL | CEL-HYB1 | 2.3 (1.2–4.4); | 5.2 (3.2–8.5); | Directly taken from Fjeld et al. [41] | D8 | ||||
| p = 0.016 | p = 1.2 × 10−11 | ||||||||
| CTRB1-CTRB2 | rs8055167 | 1.35 (1.23–1.60) | 1.09 (0.82–1.44) | Directly taken from Rosendahl et al. [42] | D1 |
| Gene | Variant | Number of Alcoholic Controls/Normal Controls | Allele Frequency in Alcoholic Controls | Allele Frequency in Normal Controls | p Value | Source of Data |
|---|---|---|---|---|---|---|
| PRSS1 | rs10273639 (C is the risk allele) | 1530/2825 | 57.7% (1766/3060) | 58.5% (3305/5650) | 0.85 | Derikx et al. [40] (only data from the German subjects were used) |
| SPINK1 | c.101A>G (p.Asn34Ser) | 305/941 | 0.49% (3/610) | 0.43% (8/1882) | 0.91 | Meta-analysis of four studies (refer to Figure S6). |
| CTRC | Aggregate pathogenic rare/very rare variants in exons 2, 3 and 7 | 432/2804 | 0.46% (4/864) | 0.45% (25/5608) | 1.00 | Rosendahl et al. [10] |
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Primers 2017, 3, 17060. [Google Scholar] [CrossRef]
- Singhvi, A.; Yadav, D. Myths and realities about alcohol and smoking in chronic pancreatitis. Curr. Opin. Gastroenterol. 2018, 34, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Tóth, M. Genetics, cell biology, and pathophysiology of pancreatitis. Gastroenterology 2019, 156, 1951–1968.e1. [Google Scholar] [CrossRef]
- Hegyi, P.; Párniczky, A.; Lerch, M.M.; Sheel, A.R.; Rebours, V.; Forsmark, C.E.; Del Chiaro, M.; Rosendahl, J.; De-Madaria, E.; Szücs, Á.; et al. International consensus guidelines for risk factors in chronic pancreatitis. Recommendations from the Working Group for the International Consensus Guidelines for Chronic Pancreatitis in collaboration with the International Association of Pancreatology, the American Pancreatic Association, the Japan Pancreas Society, and European Pancreatic Club. Pancreatology 2020, 20, 579–585. [Google Scholar] [CrossRef]
- Whitcomb, D.C.; Gorry, M.C.; Preston, R.; Furey, W.; Sossenheimer, M.J.; Ulrich, C.D.; Martin, S.P.; Gates, L.K., Jr.; Amann, S.T.; Toskes, P.P.; et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 1996, 14, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Luck, W.; Hennies, H.C.; Classen, M.; Kage, A.; Lass, U.; Landt, O.; Becker, M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 2000, 25, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Luck, W.; Becker, M.; Böhmig, M.; Kage, A.; Truninger, K.; Ammann, R.W.; O’Reilly, D.; Kingsnorth, A.; Schulz, H.-U.; et al. Mutation in the SPINK1 trypsin inhibitor gene, alcohol use, and chronic pancreatitis. JAMA 2001, 285, 2716–2717. [Google Scholar] [CrossRef]
- Aoun, E.; Chang, C.-C.H.; Greer, J.B.; Papachristou, G.I.; Barmada, M.M.; Whitcomb, D.C. Pathways to injury in chronic pancreatitis: Decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS ONE 2008, 3, e2003. [Google Scholar] [CrossRef]
- Di Leo, M.; Bianco, M.; Zuppardo, R.A.; Guslandi, M.; Calabrese, F.; Mannucci, A.; Neri, T.M.; Testoni, P.A.; Leandro, G.; Cavestro, G.M. Meta-analysis of the impact of SPINK1 p.N34S gene variation in Caucasic patients with chronic pancreatitis. An update. Dig. Liver Dis. 2017, 49, 847–853. [Google Scholar] [CrossRef]
- Rosendahl, J.; Witt, H.; Szmola, R.; Bhatia, E.; Ózsvári, B.; Landt, O.; Schulz, H.-U.; Gress, T.M.; Pfutzer, R.H.; Löhr, M.; et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 2008, 40, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Beer, S.; Rosendahl, J.; Chen, J.-M.; Chandak, G.R.; Masamune, A.; Bence, M.; Szmola, R.; Oracz, G.; Macek, M.J.; et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 2013, 45, 1216–1220. [Google Scholar] [CrossRef]
- Whitcomb, D.C.; Alzheimer’s Disease Genetics Consortium; LaRusch, J.; Krasinskas, A.M.; Klei, L.; Smith, J.P.; Brand, R.E.; Neoptolemos, J.P.; Lerch, M.M.; Tector, M.; et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat. Genet. 2012, 44, 1349–1354. [Google Scholar] [CrossRef]
- Herzig, A.F.; Génin, E.; Cooper, D.N.; Masson, E.; Férec, C.; Chen, J.-M. Role of the common PRSS1-PRSS2 haplotype in alcoholic and non-alcoholic chronic pancreatitis: Meta- and re-analyses. Genes 2020, 11, 1349. [Google Scholar] [CrossRef] [PubMed]
- Genetic Risk Factors in Chronic Pancreatitis. Available online: http://www.Pancreasgenetics.Org/index.Php (accessed on 16 February 2021).
- Le Maréchal, C.; Masson, E.; Chen, J.-M.; Morel, F.; Ruszniewski, P.; Levy, P.; Férec, C. Hereditary pancreatitis caused by triplication of the trypsinogen locus. Nat. Genet. 2006, 38, 1372–1374. [Google Scholar] [CrossRef]
- Masson, E.; Le Maréchal, C.; Chandak, G.R.; Lamoril, J.; Bezieau, S.; Mahurkar, S.; Bhaskar, S.; Reddy, D.N.; Chen, J.-M.; Férec, C. Trypsinogen copy number mutations in patients with idiopathic chronic pancreatitis. Clin. Gastroenterol. Hepatol. 2008, 6, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Masson, E.; Chen, J.M.; Scotet, V.; Le Maréchal, C.; Férec, C. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum. Genet. 2008, 123, 83–91. [Google Scholar] [CrossRef]
- Szabó, A.; Toldi, V.; Gazda, L.D.; Demcsák, A.; Tőzsér, J.; Sahin-Tóth, M. Defective binding of SPINK1 variants is an uncommon mechanism for impaired trypsin inhibition in chronic pancreatitis. J. Biol. Chem. 2021, 296, 100343. [Google Scholar] [CrossRef]
- Beer, S.; Zhou, J.; Szabó, A.; Keiles, S.; Chandak, G.R.; Witt, H.; Sahin-Tóth, M. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 2013, 62, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, E.; Sahin-Tóth, M. Genetic risk in chronic pancreatitis: The trypsin-dependent pathway. Dig. Dis. Sci. 2017, 62, 1692–1701. [Google Scholar] [CrossRef]
- Masson, E.; Chen, J.-M.; Cooper, D.N.; Férec, C. PRSS1 copy number variants and promoter polymorphisms in pancreatitis: Common pathogenetic mechanism, different genetic effects. Gut 2018, 67, 592–593. [Google Scholar] [CrossRef]
- Gui, F.; Zhang, Y.; Wan, J.; Zhan, X.; Yao, Y.; Li, Y.; Haddock, A.N.; Shi, J.; Guo, J.; Chen, J.; et al. Trypsin activity governs increased susceptibility to pancreatitis in mice expressing human PRSS1R122H. J. Clin. Investig. 2020, 130, 189–202. [Google Scholar] [CrossRef]
- Huang, H.; Swidnicka-Siergiejko, A.K.; Daniluk, J.; Gaiser, S.; Yao, Y.; Peng, L.; Zhang, Y.; Liu, Y.; Dong, M.; Zhan, X.; et al. Transgenic expression of PRSS1R122H sensitizes mice to pancreatitis. Gastroenterology 2020, 158, 1072–1082.e7. [Google Scholar] [CrossRef] [PubMed]
- Jancsó, Z.; Sahin-Tóth, M. Mutation that promotes activation of trypsinogen increases severity of secretagogue-induced pancreatitis in mice. Gastroenterology 2020, 158, 1083–1094. [Google Scholar] [CrossRef]
- Wan, J.; Haddock, A.; Edenfield, B.; Ji, B.; Bi, Y. Transgenic expression of human PRSS2 exacerbates pancreatitis in mice. Gut 2020, 69, 2051–2052. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 16 February 2021).
- Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, T.C.C. 2014. Available online: https://review-manager.software.informer.com/5.3/ (accessed on 16 February 2021).
- Sedgwick, P.; Marston, L. How to read a funnel plot in a meta-analysis. BMJ 2015, 351, h4718. [Google Scholar] [CrossRef]
- Sterne, J.A.C.; Sutton, A.J.; Ioannidis, J.P.A.; Terrin, N.; Jones, D.R.; Lau, J.; Carpenter, J.; Rücker, G.; Harbord, R.M.; Schmid, C.H.; et al. Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomised controlled trials. BMJ 2011, 343, d4002. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- PROSPERO (International Prospective Register of Systematic Reviews). Available online: https://www.Crd.York.Ac.Uk/prospero/ (accessed on 16 February 2021).
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- LaRusch, J.; Lozano-Leon, A.; Stello, K.; Moore, A.; Muddana, V.; O’Connell, M.; Diergaarde, B.; Yadav, D.; Whitcomb, D.C. The common chymotrypsinogen C (CTRC) variant G60G (c.180T) increases risk of chronic pancreatitis but not recurrent acute pancreatitis in a North American population. Clin. Transl. Gastroenterol. 2015, 6, e68. [Google Scholar] [CrossRef]
- Zou, W.-B.; Tang, X.-Y.; Zhou, D.-Z.; Qian, Y.-Y.; Hu, L.-H.; Yu, F.-F.; Yu, D.; Wu, H.; Deng, S.-J.; Lin, J.-H.; et al. SPINK1, PRSS1, CTRC, and CFTR genotypes influence disease onset and clinical outcomes in chronic pancreatitis. Clin. Transl. Gastroenterol. 2018, 9, 204. [Google Scholar] [CrossRef]
- Schneider, A.; Pfützer, R.H.; Barmada, M.M.; Slivka, A.; Martin, J.; Whitcomb, D.C. Limited contribution of the SPINK1 N34S mutation to the risk and severity of alcoholic chronic pancreatitis: A report from the United States. Dig. Dis. Sci. 2003, 48, 1110–1115. [Google Scholar] [CrossRef]
- Cichoż-Lach, H.; Michalak, M.; Lis, E.; Wojcierowski, J.; Kowalik, A.; Słomka, M.; Korolczuk, A. The N34S mutation of the SPINK1 gene and alcoholic chronic pancreatitis. Pol. Arch. Med. Wewn. 2012, 122, 277–283. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Madro, A.; Ciesielka, M.; Celinski, K.; Slomka, M.; Czechowska, G.; Kurzepa, J.; Kaszelan-Szczerbinska, B.; Buszewicz, G.; Madro, R. The genetic predisposition and its impact on the diabetes mellitus development in patients with alcoholic chronic pancreatitis. Gastroenterol. Res. Pract. 2015, 2015, 309156. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cichoz-Lach, H.; Michalak-Wojnowska, M.; Lis-Janczarek, E.; Wojcierowski, J.; Hydzik, M. Do CTRC mutations affect the development of alcoholic chronic pancreatitis and its course among poles: Preliminary study. Adv. Clin. Exp. Med. 2019, 28, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.D.; Talluri, J.; Wilcox, C.M.; Abberbock, J.N.; Tang, G.; Conwell, D.L.; Banks, P.A.; Cote, G.A.; Sherman, S.; Alkaade, S.; et al. Differences in age at onset of symptoms, and effects of genetic variants, in patients with early vs late-onset idiopathic chronic pancreatitis in a North American cohort. Clin. Gastroenterol. Hepatol. 2021, 19, 349–357. [Google Scholar] [CrossRef]
- Derikx, M.H.; Kovacs, P.; Scholz, M.; Masson, E.; Chen, J.-M.; Ruffert, C.; Lichtner, P.; Morsche, R.H.M.T.; Cavestro, G.M.; Férec, C.; et al. Polymorphisms at PRSS1–PRSS2 and CLDN2–MORC4 loci associate with alcoholic and non-alcoholic chronic pancreatitis in a European replication study. Gut 2015, 64, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Fjeld, K.; Weiss, F.U.; Lasher, D.; Rosendahl, J.; Chen, J.-M.; Johansson, B.B.; Kirsten, H.; Ruffert, C.; Masson, E.; Steine, S.J.; et al. A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat. Genet. 2015, 47, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, J.; Kirsten, H.; Hegyi, E.; Kovacs, P.; Weiss, F.U.; Laumen, H.; Lichtner, P.; Ruffert, C.; Chen, J.-M.; Masson, E.; et al. Genome-wide association study identifies inversion in the CTRB1-CTRB2 locus to modify risk for alcoholic and non-alcoholic chronic pancreatitis. Gut 2018, 67, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Schnúr, A.; Beer, S.; Witt, H.; Hegyi, P.; Sahin-Tóth, M. Functional effects of 13 rare PRSS1 variants presumed to cause chronic pancreatitis. Gut 2014, 63, 337–343. [Google Scholar] [CrossRef]
- Boulling, A.; Sato, M.; Masson, E.; Génin, E.; Chen, J.-M.; Férec, C. Identification of a functional PRSS1 promoter variant in linkage disequilibrium with the chronic pancreatitis-protecting rs10273639. Gut 2015, 64, 1837–1838. [Google Scholar] [CrossRef]
- Boulling, A.; Masson, E.; Zou, W.; Paliwal, S.; Wu, H.; Issarapu, P.; Bhaskar, S.; Génin, E.; Cooper, D.N.; Li, Z.; et al. Identification of a functional enhancer variant within the chronic pancreatitis-associated SPINK1 c.101A>G (p.Asn34Ser)-containing haplotype. Hum. Mutat. 2017, 38, 1014–1024. [Google Scholar] [CrossRef]
- Hegyi, E.; Sahin-Tóth, M. Human CPA1 mutation causes digestive enzyme misfolding and chronic pancreatitis in mice. Gut 2019, 68, 301–312. [Google Scholar] [CrossRef]
- Cassidy, B.M.; Zino, S.; Fjeld, K.; Molven, A.; Lowe, M.E.; Xiao, X. Single nucleotide polymorphisms in CEL-HYB1 increase risk for chronic pancreatitis through proteotoxic misfolding. Hum. Mutat. 2020, 41, 1967–1978. [Google Scholar] [CrossRef]
- Sahin-Tóth, M. Genetic risk in chronic pancreatitis: The misfolding-dependent pathway. Curr. Opin. Gastroenterol. 2017, 33, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Orekhova, A.; Geisz, A.; Sahin-Tóth, M. Ethanol feeding accelerates pancreatitis progression in CPA1 N256K mutant mice. Am. J. Physiol. Gastrointest Liver Physiol. 2020, 318, G694–G704. [Google Scholar] [CrossRef]
- Lankisch, M.R.; Imoto, M.; Layer, P.; DiMagno, E.P. The Effect of small amounts of alcohol on the clinical course of chronic pancreatitis. Mayo Clin. Proc. 2001, 76, 242–251. [Google Scholar] [CrossRef]
- Zou, W.-B.; Boulling, A.; Masamune, A.; Issarapu, P.; Masson, E.; Wu, H.; Sun, X.-T.; Hu, L.-H.; Zhou, D.-Z.; He, L.; et al. No association between CEL–HYB hybrid allele and chronic pancreatitis in Asian populations. Gastroenterology 2016, 150, 1558–1560.e5. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-Y.; Zou, W.-B.; Masson, E.; Hu, L.-H.; Férec, C.; Chen, J.-M.; Li, Z.-S.; Liao, Z. The CTRB1-CTRB2 risk allele for chronic pancreatitis discovered in European populations does not contribute to disease risk variation in the Chinese population due to near allele fixation. Gut 2018, 67, 1368–1369. [Google Scholar] [CrossRef]
- Lasher, D.; Szabó, A.; Masamune, A.; Chen, J.-M.; Xiao, X.; Whitcomb, D.C.; Barmada, M.M.; Ewers, M.; Ruffert, C.; Paliwal, S.; et al. Protease-sensitive pancreatic lipase variants are associated with early onset chronic pancreatitis. Am. J. Gastroenterol. 2019, 114, 974–983. [Google Scholar] [CrossRef] [PubMed]


| Gene | Variant | Number of ACP/NACP/Normal Controls | Allele Frequency in ACP | Allele Frequency in NACP | Allele Frequency in Normal Controls | ORACP (95% CI); p Value | ORNACP (95% CI); p Value | Source of Data | Data Point Denoted in Figure 2 |
|---|---|---|---|---|---|---|---|---|---|
| PRSS1 | rs10273639C/T (C is the risk allele) | 65.0% | 53.6% | 1.67 (1.56–1.78); | 1.28 (1.17–1.40); | Directly taken from Herzig et al. [13] | D3 | ||
| (3610/5556) | (4341/8094) | p < 0.00001 | p < 0.00001 | ||||||
| c.365G>A (p.Arg122His) | 719/1185/2330 | 0.7% | 0.9% | 0.1% | 7.01 (1.83–26.77); | 10.22 (3.52–29.70); | Meta-analysis of 12 studies (refer to Figure S2) | D10 | |
| 10/1438 | 22/2370 | 3/4660 | p = 0.004 | p < 0.0001 | |||||
| c.623G>C (p.Gly208Ala) | 206/715/1196 | 4.4% | 4.2% | 0.9% | 4.92 (2.62–9.26); | 4.72 (2.88–7.72); p = 9.2 × 10−11 | Zou et al. [34] | D7 | |
| (18/412) | (60/1430) | (22/2392) | p = 3.6 × 10−7 | ||||||
| SPINK1 | c.101A>G (p.Asn34Ser) | 1184/1441/4021 | 3.0% | 5.8% | 0.6% | 4.55 (3.08–6.72); | 10.90 (7.56–15.72); | Meta-analysis of 17 studies (refer to Figure S3) | D11 |
| (72/2368) | (168/2882) | (49/8042) | p < 0.00001 | p < 0.00001 | |||||
| c.194+2T>C | 206/715/1196 | 14.3% | 24.5% | 0.5% | 30.59 (16.61–56.34); | 59.31 (33.93–103.64); | Zou et al. [34] | D13 | |
| (59/412) | (350/1430) | (13/2392) | p < 2.2 × 10−16 | p < 2.2 × 10−16 | |||||
| CTRC | Aggregate rare/very rare pathogenic variants in exons 2, 3 and 7 | 348/758/2804 | 1.9% | 1.8% | 0.4% | 4.25 (2.16–8.35); | 4.05 (2.34–7.00); | Rosendahl et al. [10] | D6 |
| (13/696) | (27/1516) | (25/5608) | p = 2.0 × 10−5 | p = 2.1 × 10−7 | |||||
| c.760C>T (p.Arg254Trp) | 788/1563/4349 | 0.8% | 0.4% | 0.3% | 2.87 (1.34–6.14); | 1.98 (1.03–3.81); | Meta-analysis of four studies (refer to Figure S5) | D5 | |
| (12/1576) | (13/3126) | (22/8698) | p = 0.007 | p = 0.04 | |||||
| c.180C>T (p.Gly60Gly) | 236/302/1013 | 20.8% | 12.4% | 10.8% | 2.16 (1.66–2.81); | 1.17 (0.89–1.55); | LaRush et al. [33] | D2 | |
| (98/472) | (75/604) | (219/2026) | p = 7.7 × 10−7 | p = 0.36 | |||||
| c.180C>T (p.Gly60Gly) | 206/715/1196 | 0.5% | 1.1% | 0.1% | 3.88 (0.65–23.32); | 9.01 (2.62–30.98); | Zou et al. [34] | D9 | |
| (2/412) | (16/1430) | (3/2392) | p = 0.33 | p = 7.4 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-M.; Herzig, A.F.; Génin, E.; Masson, E.; Cooper, D.N.; Férec, C. Scale and Scope of Gene-Alcohol Interactions in Chronic Pancreatitis: A Systematic Review. Genes 2021, 12, 471. https://doi.org/10.3390/genes12040471
Chen J-M, Herzig AF, Génin E, Masson E, Cooper DN, Férec C. Scale and Scope of Gene-Alcohol Interactions in Chronic Pancreatitis: A Systematic Review. Genes. 2021; 12(4):471. https://doi.org/10.3390/genes12040471
Chicago/Turabian StyleChen, Jian-Min, Anthony F. Herzig, Emmanuelle Génin, Emmanuelle Masson, David N. Cooper, and Claude Férec. 2021. "Scale and Scope of Gene-Alcohol Interactions in Chronic Pancreatitis: A Systematic Review" Genes 12, no. 4: 471. https://doi.org/10.3390/genes12040471
APA StyleChen, J.-M., Herzig, A. F., Génin, E., Masson, E., Cooper, D. N., & Férec, C. (2021). Scale and Scope of Gene-Alcohol Interactions in Chronic Pancreatitis: A Systematic Review. Genes, 12(4), 471. https://doi.org/10.3390/genes12040471