
The Conservation of Low Complexity Regions in Bacterial Proteins Depends on the Pathogenicity of the Strain and Subcellular Location of the Protein



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Retrieval
2.2. Orthology Assessment
2.3. Conservation of Protein Features
2.4. Subcellular Location Prediction
3. Results
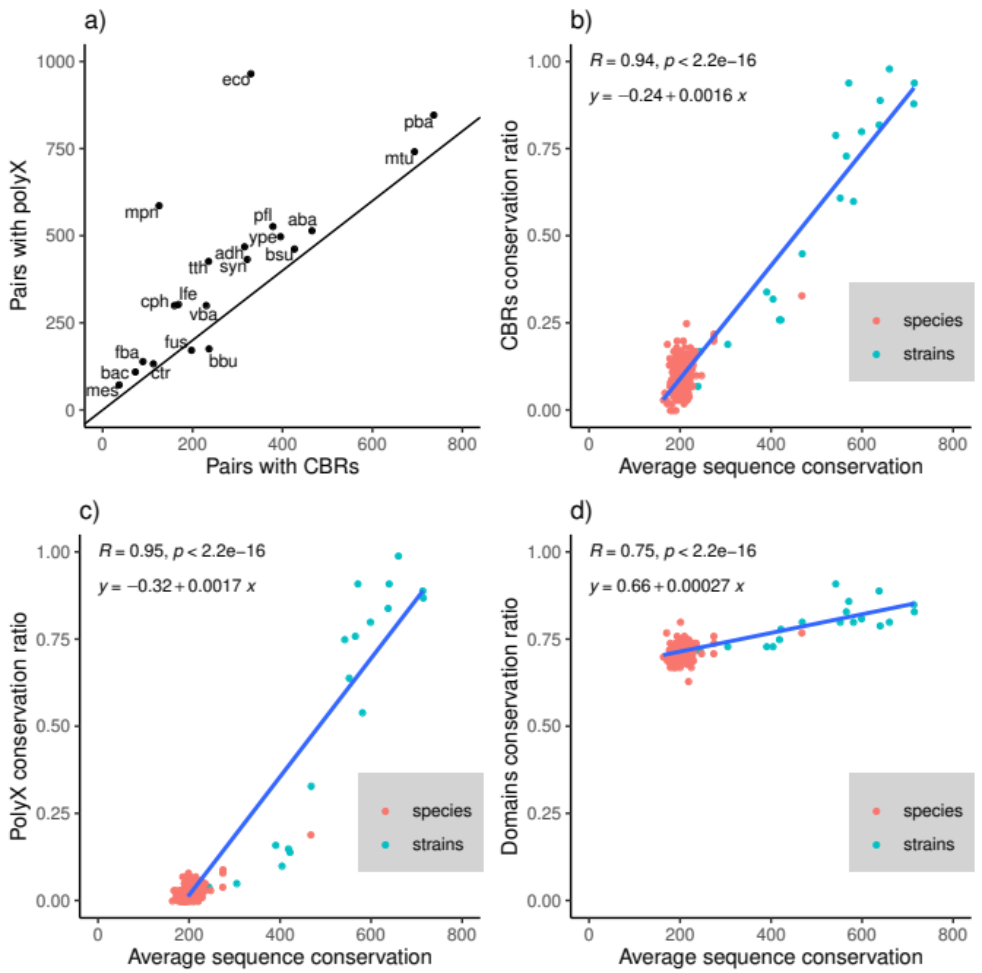
3.1. Conservation of LCRs in Bacterial Strains
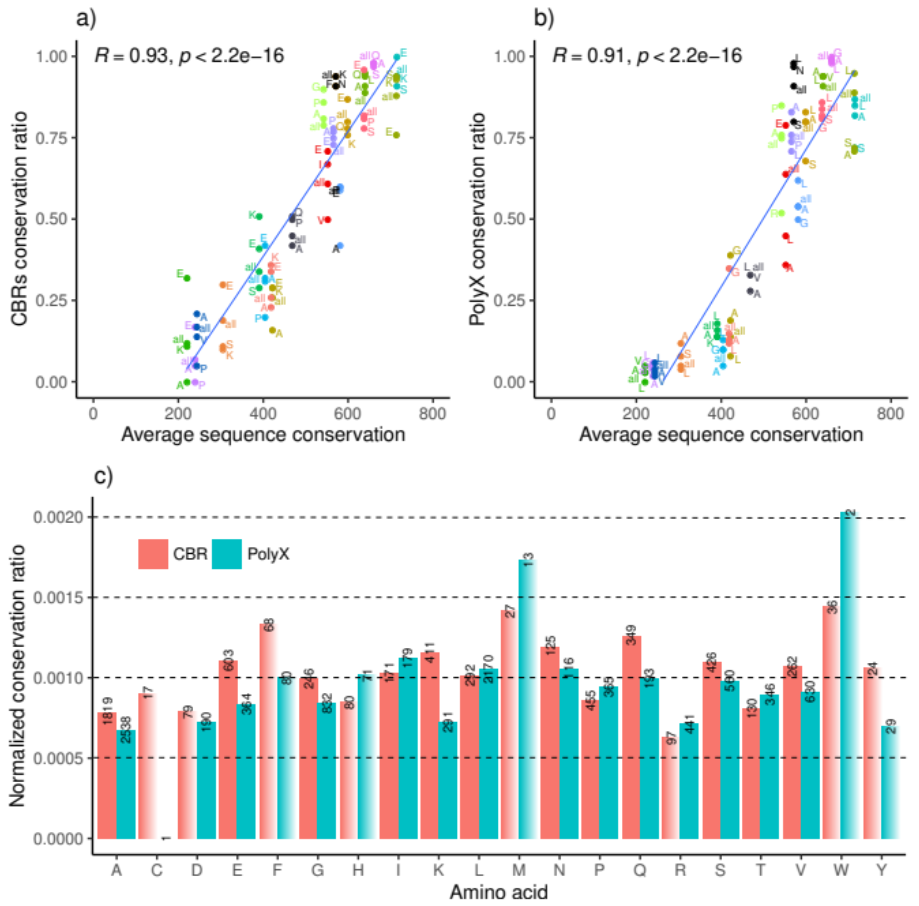
3.2. LCR Conservation Patterns and Amino Acid Type
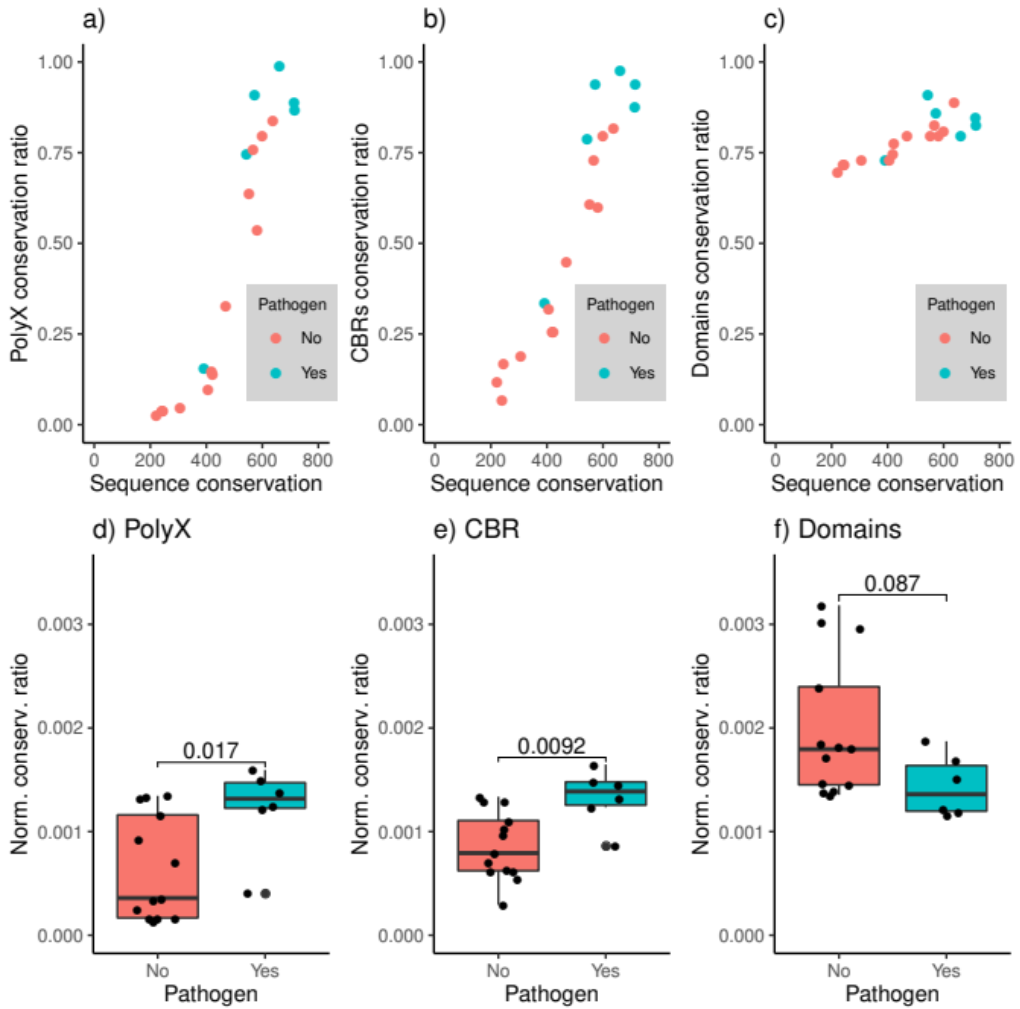
3.3. LCRs Are More Conserved in Pathogenic Strain Pairs
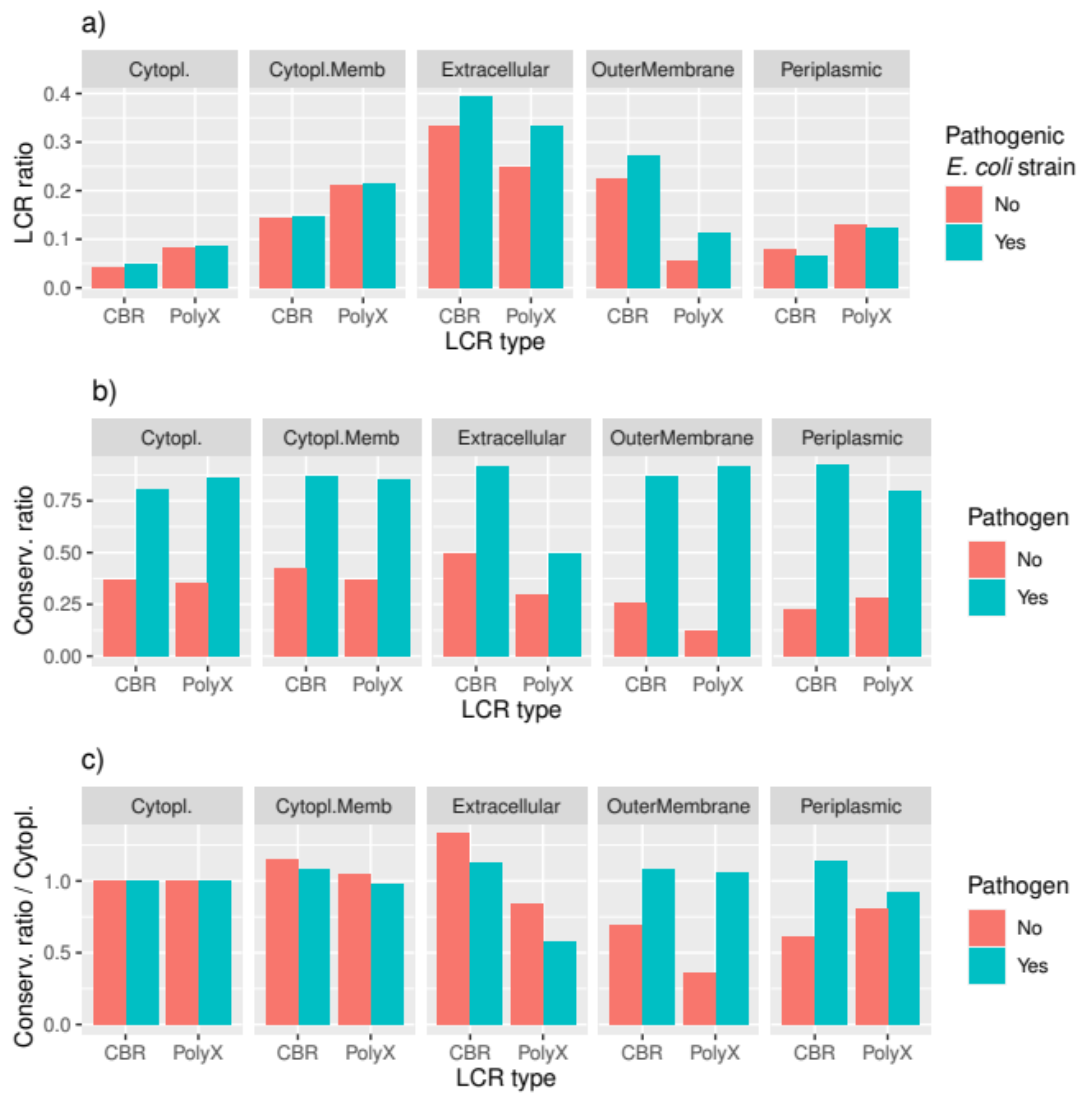
3.4. Conservation of LCRs Is Different in the Extracellular and Outer Membrane Proteins from Pathogens
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mier, P.; Andrade-Navarro, M.A. Assessing the low complexity of protein sequences via the low complexity triangle. PLoS ONE 2020, 15, e0239154. [Google Scholar] [CrossRef]
- Lenz, C.; Haerty, W.; Golding, G.B. Increased substitution rates surrounding low-complexity regions within primate proteins. Genome Biol. Evol. 2014, 6, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef]
- Babu, M.M.; Kriwacki, R.W.; Pappu, R.V. Structural biology. Versatility from protein disorder. Science 2012, 337, 1460–1461. [Google Scholar] [CrossRef]
- Sim, K.L.; Creamer, T.P. Abundance and distributions of eukaryote protein simple sequences. Mol. Cell. Proteom. 2002, 1, 983–995. [Google Scholar] [CrossRef]
- Ntountoumi, C.; Vlastaridis, P.; Mossialos, D.; Stathopoulos, C.; Iliopoulos, I.; Promponas, V.; Oliver, S.G.; Amoutzias, G.D. Low complexity regions in the proteins of prokaryotes perform important functional roles and are highly conserved. Nucleic Acids Res. 2019, 47, 9998–10009. [Google Scholar] [CrossRef]
- Klemm, P.; Schembri, M.A. Bacterial adhesins: Function and structure. Int. J. Med. Microbiol. 2000, 290, 27–35. [Google Scholar] [CrossRef]
- Dijkshoorn, L.; Ursing, B.M.; Ursing, J.B. Strain, clone and species: Comments on three basic concepts of bacteriology. J. Med. Microbiol. 2000, 49, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G.; Brenner, D.J.; Colwell, R.R.; Grimont, P.A.D.; Kandler, O.; Krichevsky, M.I.; Moore, L.H.; Moore, W.E.C.; Murray, R.; Stackebrandt, E.S.M.P.; et al. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Bact. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, W.F. Population genomics: How bacterial species form and why they don’t exist. Curr. Biol. 2012, 22, R451–R453. [Google Scholar] [CrossRef]
- Edwards, S.V.; Potter, S.; Schmitt, C.J.; Bragg, J.G.; Moritz, C. Reticulation, divergence, and the phylogeography-phylogenetics continuum. Proc. Natl. Acad. Sci. USA 2016, 113, 8025–8032. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, T.; Ferretti, P.; Maistrenko, O.M.; Bork, P. Diversity within species: Interpreting strains in microbiomes. Nat. Rev. Microbiol. 2020, 18, 491–506. [Google Scholar] [CrossRef]
- Coates, D.J.; Byrne, M.; Moritz, C. Genetic diversity and conservation units: Dealing with the species-population continuum in the age of genomics. Front. Ecol. Evol. 2018, 6. [Google Scholar] [CrossRef]
- Roux, C.; Fraïsse, C.; Romiguier, J.; Anciaux, Y.; Galtier, N.; Bierne, N. Shedding Light on the Grey Zone of Speciation along a Continuum of Genomic Divergence. PLoS Biol. 2016, 14, e2000234. [Google Scholar] [CrossRef]
- UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database (Oxford) 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Mier, P.; Elena-Real, C.; Urbanek, A.; Bernadó, P.; Andrade-Navarro, M.A. The importance of definitions in the study of polyQ regions: A tale of thresholds, impurities and sequence context. Comput. Struct. Biotechnol. J. 2020, 18, 306–313. [Google Scholar] [CrossRef]
- Jarnot, P.; Ziemska-Legiecka, J.; Dobson, L.; Merski, M.; Mier, P.; Andrade-Navarro, M.A.; Hancock, J.M.; Dosztányi, Z.; Paladin, L.; Necci, M.; et al. PlaToLoCo: The first web meta-server for visualization and annotation of low complexity regions in proteins. Nucleic Acids Res. 2020, 48, W77–W84. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Mier, P.; Paladin, L.; Tamana, S.; Petrosian, S.; Hajdu-Soltész, B.; Urbanek, A.; Gruca, A.; Plewczynski, D.; Grynberg, M.; Bernadó, P.; et al. Disentangling the complexity of low complexity proteins. Brief. Bioinform. 2020, 21, 458–472. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Ramette, A.; Tiedje, J.M. The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1929–1940. [Google Scholar] [CrossRef]
- Caro-Quintero, A.; Konstantinidis, K.T. Bacterial species may exist, metagenomics reveal. Environ. Microbiol. 2012, 14, 347–355. [Google Scholar] [CrossRef]
- Pelassa, I.; Fiumara, F. Differential Occurrence of Interactions and Interaction Domains in Proteins Containing Homopolymeric Amino Acid Repeats. Front. Genet. 2015, 6, 345. [Google Scholar] [CrossRef]
- Schaefer, M.H.; Wanker, E.E.; Andrade-Navarro, M.A. Evolution and function of CAG/polyglutamine repeats in protein-protein interaction networks. Nucleic Acids Res. 2012, 40, 4273–4287. [Google Scholar] [CrossRef]
- Diard, M.; Hardt, W.D. Evolution of bacterial virulence. FEMS Microbiol. Rev. 2017, 41, 679–697. [Google Scholar] [CrossRef]
- Brites, D.; Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol. Rev. 2015, 264, 6–24. [Google Scholar] [CrossRef]
- Sheldon, I.M.; Rycroft, A.N.; Dogan, B.; Craven, M.; Bromfield, J.J.; Chandler, A.; Roberts, M.H.; Price, S.B.; Gilbert, R.O.; Simpson, K.W. Specific strains of Escherichia coli are pathogenic for the endometrium of cattle and cause pelvic inflammatory disease in cattle and mice. PLoS ONE 2010, 5, e9192. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, J.; Yao, H.; Hu, H. Fusobacterium and Colorectal Cancer. Front. Oncol. 2018, 8, 371. [Google Scholar] [CrossRef]
- Karpiński, T.M. Role of Oral Microbiota in Cancer Development. Microorganisms 2019, 7, 20. [Google Scholar] [CrossRef]
- Moxon, R.; Bayliss, C.; Hood, D. Bacterial contingency loci: The role of simple sequence DNA repeats in bacterial adaptation. Annu. Rev. Genet. 2006, 40, 307–333. [Google Scholar] [CrossRef]
- Hou, N.; Jiang, N.; Ma, Y.; Zou, Y.; Piao, X.; Liu, S.; Chen, Q. Low-Complexity Repetitive Epitopes of Plasmodium falciparum Are Decoys for Humoural Immune Responses. Front. Immunol. 2020, 11, 610. [Google Scholar] [CrossRef] [PubMed]
- Totzeck, F.; Andrade-Navarro, M.A.; Mier, P. The Protein Structure Context of PolyQ Regions. PLoS ONE 2017, 12, e0170801. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mier, P.; Andrade-Navarro, M.A. The Conservation of Low Complexity Regions in Bacterial Proteins Depends on the Pathogenicity of the Strain and Subcellular Location of the Protein. Genes 2021, 12, 451. https://doi.org/10.3390/genes12030451
Mier P, Andrade-Navarro MA. The Conservation of Low Complexity Regions in Bacterial Proteins Depends on the Pathogenicity of the Strain and Subcellular Location of the Protein. Genes. 2021; 12(3):451. https://doi.org/10.3390/genes12030451
Chicago/Turabian StyleMier, Pablo, and Miguel A. Andrade-Navarro. 2021. "The Conservation of Low Complexity Regions in Bacterial Proteins Depends on the Pathogenicity of the Strain and Subcellular Location of the Protein" Genes 12, no. 3: 451. https://doi.org/10.3390/genes12030451
APA StyleMier, P., & Andrade-Navarro, M. A. (2021). The Conservation of Low Complexity Regions in Bacterial Proteins Depends on the Pathogenicity of the Strain and Subcellular Location of the Protein. Genes, 12(3), 451. https://doi.org/10.3390/genes12030451