
FA-nf: A Functional Annotation Pipeline for Proteins from Non-Model Organisms Implemented in Nextflow

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
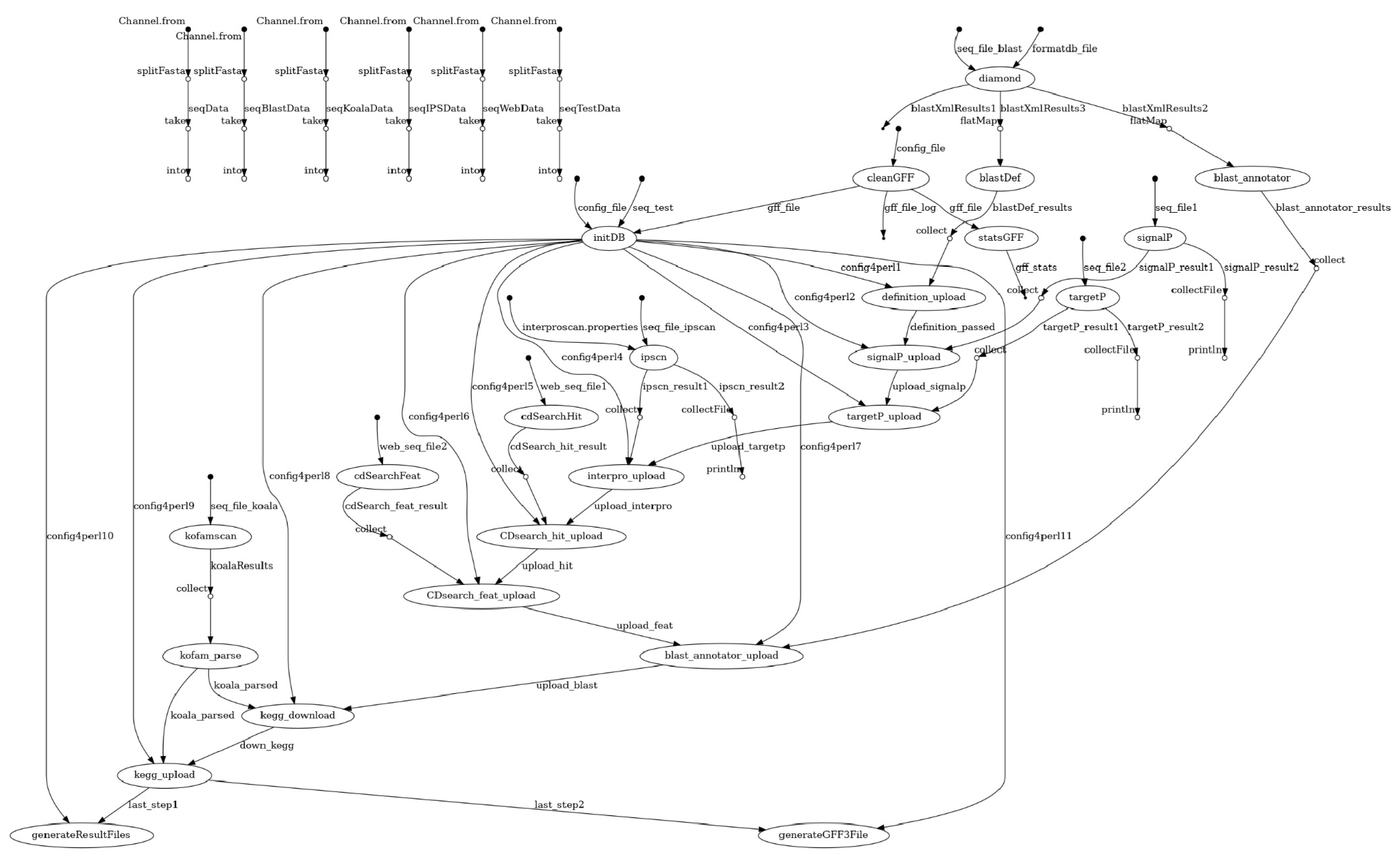
2.1. Overview of the Pipeline
2.2. Preprocessing
2.3. Analysis
2.4. NCBI BLAST+ and DIAMOND as Annotation Sources
2.5. KAAS and KOFAM
2.6. Other Programs
2.7. Integration and Reports
3. Results
3.1. Running FA-nf
- Ensure you have a recent version of Git software and clone the FA-nf repository. This will create a FA-nf folder with the pipeline contents.
- ○
- $ git clone --recursive https://github.com/guigolab/FA-nf
- You can otherwise download and extract a specific release version from the following:
- Ensure you have either Docker (at least 19.x version) or, preferably, Singularity (at least 3.2.x version) installed.
- ○
- Docker installation details: https://docs.docker.com/install/ (accessed on 19 October 2021).
- ○
- Singularity installation details: https://singularity.hpcng.org/admin-docs/3.7/installation.html (accessed on 19 October 2021).
- Install Nextflow (version 20.10.0 tested). In this example, we keep it in the same directory as the pipeline. Otherwise, you would normally place it somewhere in the PATH of your system. Java 8 or later must be available in the system.
- ○
- $ cd FA-nf; export NXF_VER=20.10.0; curl -s https://get.nextflow.io | bash
- If you plan to use Interproscan with private software, follow the container image generation instructions that can be found under the containers/interproscan directory of the repository.
- If you want to use privative programs, such as signalP and targetP, prepare a container image following the instructions under the containers/sigtarp directory of the repository. Otherwise, the execution of these applications can be skipped from the pipeline configuration.
- Optionally, you also can set up your custom GOGOApi REST API service from the instructions provided under the gogoapi directory of the repository.
- If your system does not have internet connection, you can generate Singularity files in advance and modify accordingly the container tag values in the nextflow.config file. Some pregenerated Singularity container images can be found at https://biocore.crg.eu/containers/FA-nf/ (accessed on 19 October 2021).
- Download (and index when necessary) all the datasets used by the pipeline, as detailed in the repository documentation. At least some BLAST, Interproscan and KofamKOALA files are needed.
- ○
- A Nextflow pipeline script for downloading necessary datasets (download.nf) is available. A sample configuration file (params.download.config) is available for convenience. The datasets will be downloaded and indexed using the following command:
- ■
- $ ./nextflow run -bg download.nf --config params.download.config &> download.logfile
- ○
- Alternately, some convenient scripts for setting and indexing the necessary datasets can be found at https://github.com/toniher/biomirror/
- ○
- As a last instance, some minimal test datasets can be found here: https://biocore.crg.eu/papers/FA-nf-2021/datasets/ (accessed on 19 October 2021).
- For sake of information, we provide some indicative space usage numbers below.
- ○
- NCBI databases (update_blastdb.pl) (nr, 349 G index size)
- ■
- Formatting with Diamond (nr, 187 G index size)
- ○
- Interproscan (5.48-83.0, 89 G)
- ○
- KofamKOALA ftp://ftp.genome.jp/pub/db/kofam/ (202103 > ko_list, profiles, KO text files, 13.5 G)
- ○
- Datasets used for GOGOApi retrieval service.
- ■
- UniProt ID mapping (89 G uncompressed)
- ■
- GOA. Uniprot proteins and GO accession codes mapping (11 G compressed)
- ■
- Final database size: ~250 G
- Check params.config file and adapt its values to your system configuration and to work with your input files and datasets locations as defined in previous points
- Check nextflow.config file and adjust it according to the characteristics of your HPC queue system by replacing queue names and increasing or decreasing aspects such as CPU or RAM. More details at: https://www.nextflow.io/docs/latest/config.html
- You can start the execution of the pipeline (normally from the node with access to an HPC queue system) with:
- ○
- $ ./nextflow run -bg main.nf --config params.config &> logfile
- We can check how the pipeline is progressing with:
- ○
- $ tail -f logfile
- As the pipeline advances, intermediary and final results are stored in resultPath directory, as defined in params.config file. More details can be found in the README file of the software repository.
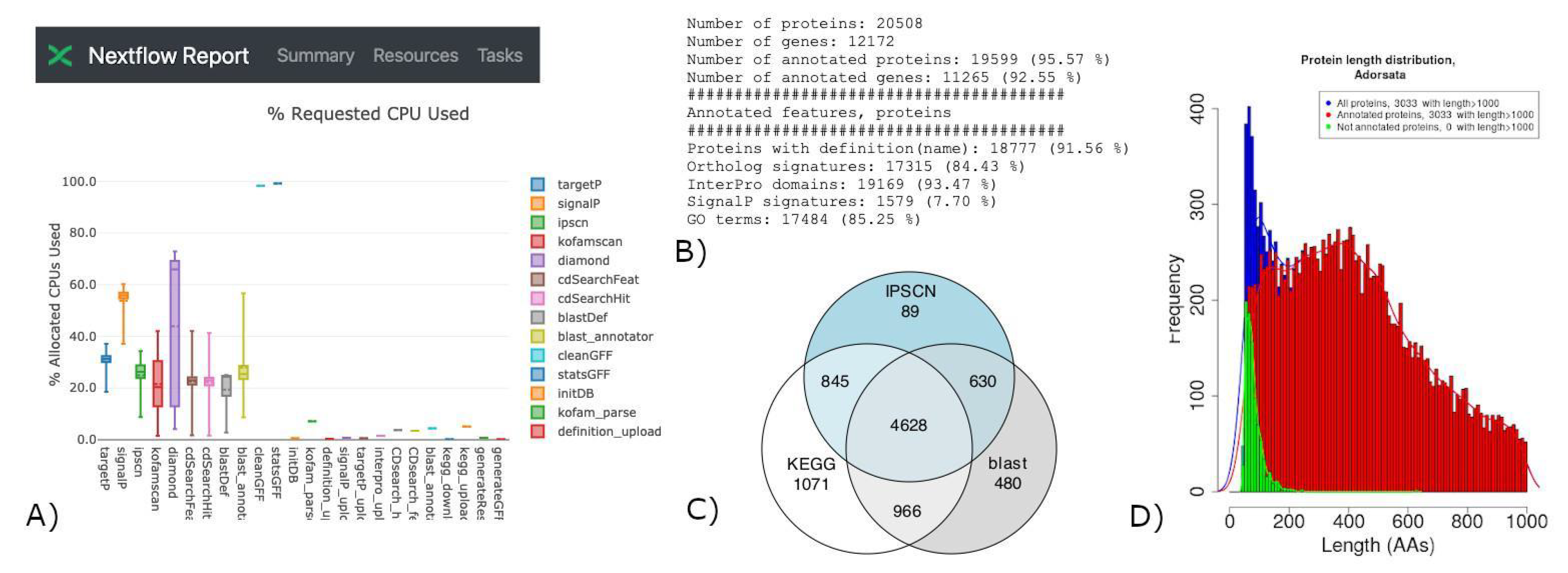
3.2. Example Cases
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2021, 49, D92–D96. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Dominguez Del Angel, V.; Hjerde, E.; Sterck, L.; Capella-Gutierrez, S.; Notredame, C.; Vinnere Pettersson, O.; Amselem, J.; Bouri, L.; Bocs, S.; Klopp, C.; et al. Ten Steps to Get Started in Genome Assembly and Annotation. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- NCBI Resource Coordinators Database. Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Galperin, M.Y.; Koonin, E.V. Sources of Systematic Error in Functional Annotation of Genomes: Domain Rearrangement, Non-Orthologous Gene Displacement and Operon Disruption. Silico Biol. 1998, 1, 55–67. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Capella-Gutiérrez, S.; Pryszcz, L.P.; Marcet-Houben, M.; Gabaldón, T. PhylomeDB v4: Zooming into the Plurality of Evolutionary Histories of a Genome. Nucleic Acids Res. 2014, 42, D897–D902. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving Coverage, Classification and Access to Protein Sequence Annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER Version 11: Expanded Annotation Data from Gene Ontology and Reactome Pathways, and Data Analysis Tool Enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Oates, M.E.; Stahlhacke, J.; Vavoulis, D.V.; Smithers, B.; Rackham, O.J.L.; Sardar, A.J.; Zaucha, J.; Thurlby, N.; Fang, H.; Gough, J. The SUPERFAMILY 1.75 Database in 2014: A Doubling of Data. Nucleic Acids Res. 2015, 43, D227–D233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humann, J.L.; Lee, T.; Ficklin, S.; Main, D. Structural and Functional Annotation of Eukaryotic Genomes with GenSAS. In Gene Prediction: Methods and Protocols; Methods in Molecular Biology; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; pp. 29–51. ISBN 978-1-4939-9173-0. [Google Scholar]
- Bryant, D.M.; Johnson, K.; Di Tommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.-H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [Green Version]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A Rapid Functional Annotation Web Server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Ruiz-Perez, C.A.; Conrad, R.E.; Konstantinidis, K.T. MicrobeAnnotator: A User-Friendly, Comprehensive Functional Annotation Pipeline for Microbial Genomes. BMC Bioinform. 2021, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Casimiro-Soriguer, C.S.; Muñoz-Mérida, A.; Pérez-Pulido, A.J. Sma3s: A Universal Tool for Easy Functional Annotation of Proteomes and Transcriptomes. Proteomics 2017, 17, 1700071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant. Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Brandies, P.A.; Hogg, C.J. Ten Simple Rules for Getting Started with Command-Line Bioinformatics. PLoS Comput. Biol. 2021, 17, e1008645. [Google Scholar] [CrossRef]
- Leipzig, J. A Review of Bioinformatic Pipeline Frameworks. Brief. Bioinform. 2017, 18, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Köster, J.; Rahmann, S. Snakemake—A Scalable Bioinformatics Workflow Engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Jalili, V.; Afgan, E.; Gu, Q.; Clements, D.; Blankenberg, D.; Goecks, J.; Taylor, J.; Nekrutenko, A. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2020 Update. Nucleic Acids Res. 2020, 48, W395–W402. [Google Scholar] [CrossRef] [PubMed]
- Cozzuto, L.; Liu, H.; Pryszcz, L.P.; Pulido, T.H.; Delgado-Tejedor, A.; Ponomarenko, J.; Novoa, E.M. Master Of Pores: A Workflow for the Analysis of Oxford Nanopore Direct RNA Sequencing Datasets. Front. Genet. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The Nf-Core Framework for Community-Curated Bioinformatics Pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Bandla, C.; Guo, J.; Vera Alvarez, R.; Bai, M.; Vizcaíno, J.A.; Moreno, P.; Grüning, B.; Sallou, O.; Perez-Riverol, Y. BioContainers Registry: Searching Bioinformatics and Proteomics Tools, Packages, and Containers. J. Proteome Res. 2021, 20, 2056–2061. [Google Scholar] [CrossRef]
- Gacek, C.; Arief, B. The Many Meanings of Open Source. IEEE Softw. 2004, 21, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.; Engelbrecht, J.; Brunak, S.; von Heijne, G. Identification of Prokaryotic and Eukaryotic Signal Peptides and Prediction of Their Cleavage Sites. Protein Eng. 1997, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Gatto, L.; Wang, R.; Sachsenberg, T.; Uszkoreit, J.; Leprevost, F.D.V.; Fufezan, C.; Ternent, T.; Eglen, S.J.; Katz, D.S.; et al. Ten Simple Rules for Taking Advantage of Git and GitHub. PLoS Comput. Biol. 2016, 15, e1007142. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinform. Oxf. Engl. 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- SQLite Frequently Asked Questions. Available online: https://www.sqlite.org/faq.html#q5 (accessed on 11 June 2021).
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific Containers for Mobility of Compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Dainat, J.; Hereñú, D.; Pucholt, P. NBISweden/AGAT: AGAT-v0.6.2; Zenodo: Geneve, Switzerland, 2021. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq: Reference Generation and Analysis with Trinity. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Camon, E.; Magrane, M.; Barrell, D.; Binns, D.; Fleischmann, W.; Kersey, P.; Mulder, N.; Oinn, T.; Maslen, J.; Cox, A.; et al. The Gene Ontology Annotation (GOA) Project: Implementation of GO in SWISS-PROT, TrEMBL, and InterPro. Genome Res. 2003, 13, 662–672. [Google Scholar] [CrossRef] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog Assignment Based on Profile HMM and Adaptive Score Threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [Green Version]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating Proteins in the Cell Using TargetP, SignalP and Related Tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The Conserved Domain Database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Tipney, H.; Hunter, L. An Introduction to Effective Use of Enrichment Analysis Software. Hum. Genom. 2010, 4, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Fouks, B.; Brand, P.; Nguyen, H.N.; Herman, J.; Camara, F.; Ence, D.; Hagen, D.; Hoff, K.J.; Nachweide, S.; Romoth, L.; et al. The Genomic Basis of Evolutionary Differentiation among Honey Bees. Genome Res. 2021, 31, gr.272310.120. [Google Scholar] [CrossRef]
- Vlasova, A.; Capella-Gutiérrez, S.; Rendón-Anaya, M.; Hernández-Oñate, M.; Minoche, A.E.; Erb, I.; Câmara, F.; Prieto-Barja, P.; Corvelo, A.; Sanseverino, W.; et al. Genome and Transcriptome Analysis of the Mesoamerican Common Bean and the Role of Gene Duplications in Establishing Tissue and Temporal Specialization of Genes. Genome Biol. 2016, 17, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Dong, Z.; Liu, G.; He, J.; Zhao, R.; Wang, W.; Peng, Y.; Li, X. Phylogenetic Analysis Provides Insights into the Evolution of Asian Fireflies and Adult Bioluminescence. Mol. Phylogenet. Evol. 2019, 140, 106600. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, K.; Imanishi, T. Human Contamination in Public Genome Assemblies. PLoS ONE 2016, 11, e0162424. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Salmerón, J.E.; Moreno-Hagelsieb, G. Progress in Quickly Finding Orthologs as Reciprocal Best Hits: Comparing Blast, Last, Diamond and MMseqs2. BMC Genom. 2020, 21, 741. [Google Scholar] [CrossRef]
- Makarewich, C.A.; Olson, E.N. Mining for Micropeptides. Trends Cell Biol. 2017, 27, 685–696. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, J.; Lian, X.; Sun, L.; Meng, K.; Chen, Y.; Sun, Z.; Yin, X.; Li, Y.; Zhao, J.; et al. A Hidden Human Proteome Encoded by “non-Coding” Genes. Nucleic Acids Res. 2019, 47, 8111–8125. [Google Scholar] [CrossRef]
- Sandve, G.K.; Nekrutenko, A.; Taylor, J.; Hovig, E. Ten Simple Rules for Reproducible Computational Research. PLoS Comput. Biol. 2013, 9, e1003285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, R.D.; Hicks, S.C. Reproducible Research: A Retrospective. Annu. Rev. Public Health 2021, 42, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, A.; Mortensen, J.M.; Winnenburg, R.; Liu, C.; Alessi, D.T.; Swamy, V.; Vallania, F.; Lofgren, S.; Haynes, W.; Shah, N.H.; et al. Interpretation of Biological Experiments Changes with Evolution of the Gene Ontology and Its Annotations. Sci. Rep. 2018, 8, 5115. [Google Scholar] [CrossRef] [PubMed]
- OpenAIRE Zenodo. European Organization For Nuclear Research; OpenAIRE Zenodo: Geneve, Switzerland, 2013. [Google Scholar] [CrossRef]
- Halchenko, Y.O.; Meyer, K.; Poldrack, B.; Solanky, D.S.; Wagner, A.S.; Gors, J.; MacFarlane, D.; Pustina, D.; Sochat, V.; Ghosh, S.S.; et al. DataLad: Distributed System for Joint Management of Code, Data, and Their Relationship. J. Open Source Softw. 2021, 6, 3262. [Google Scholar] [CrossRef]
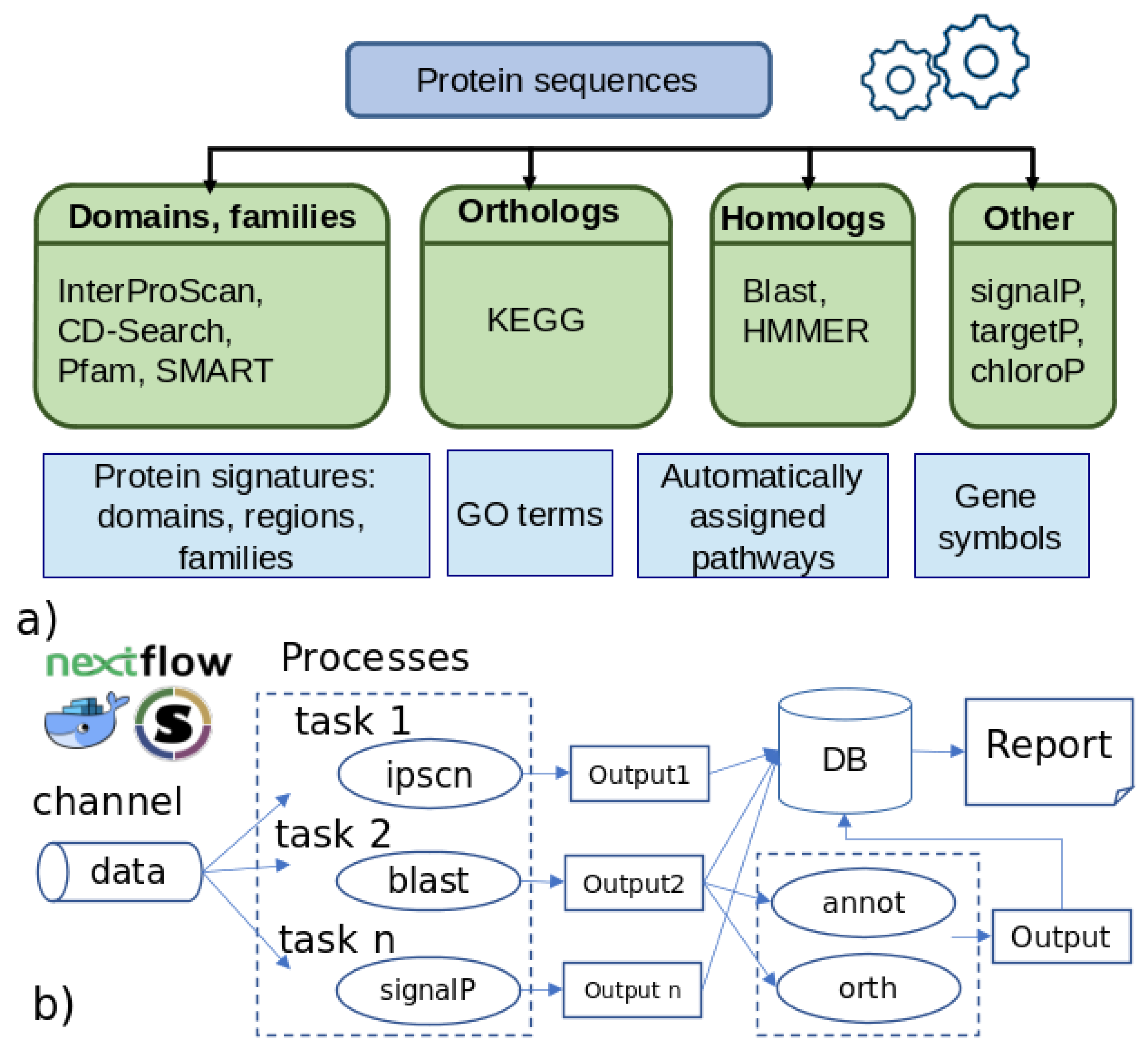
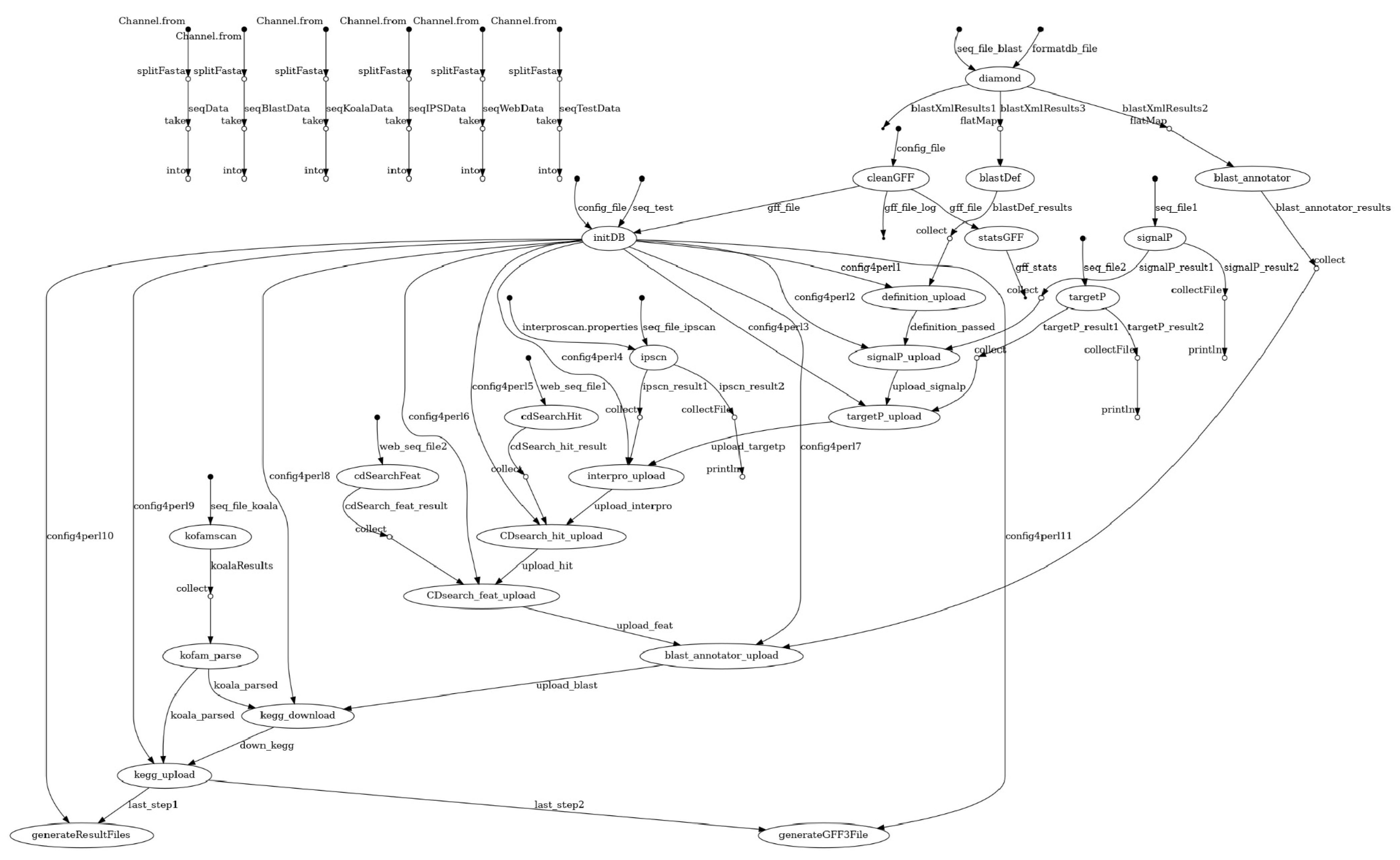
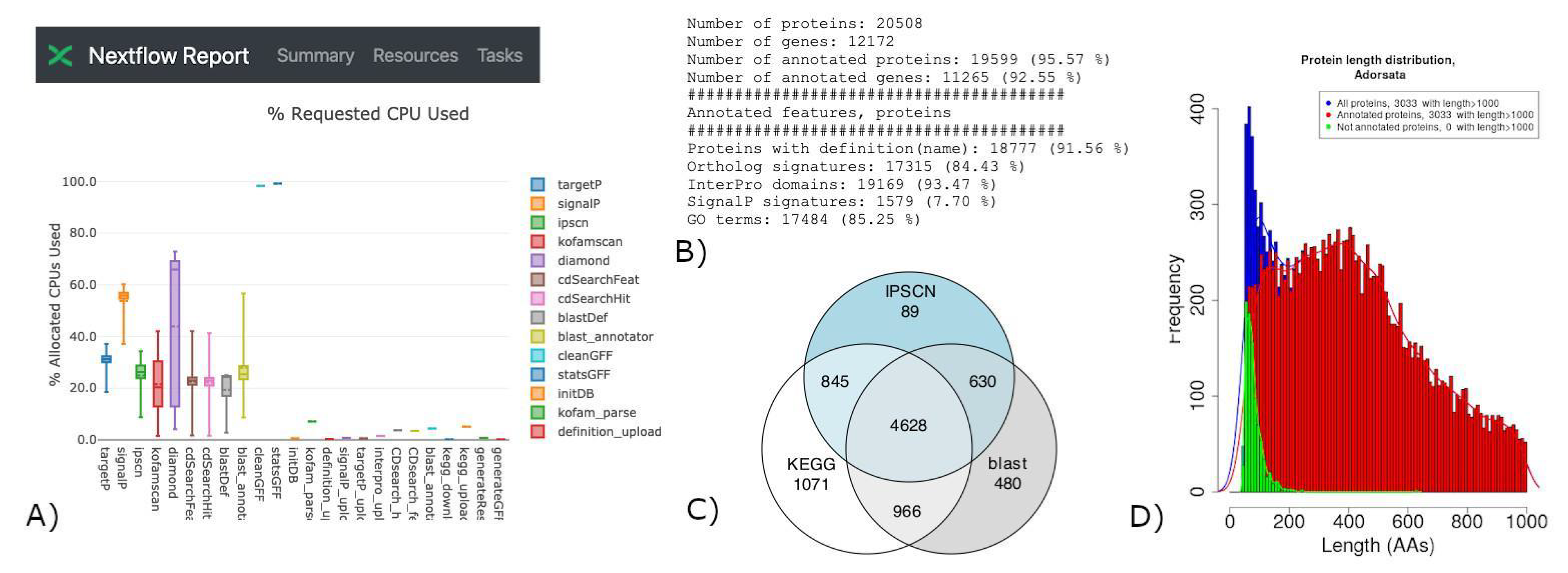

{kind=link}
{kind=link}
{kind=link}
| Program/Pipeline | Installation | Used Software | Datasets | Comments |
|---|---|---|---|---|
| Blast2GO [20] | Local installation and web/cloud services | BLAST+, Interproscan, BLAST2GO specific software, etc. | Custom, Normally, NCBI BLAST DBs, InterPro, GO | Subscription tool. Visualization dashboard. Gene structural annotation options. Newer versions integrated into other toolboxes. |
| eggNOG mapper [9] | Web service and local installation | DIAMOND, HMMER | eggNOGdb (from several sources), GO, PFAM, SMART, COG | Available command-line tool and REST API for querying the service. Gene structural annotation options. |
| FA-nf | Local installation | BLAST+, DIAMOND, Interproscan, KOFAM, CDD, SignalP, TargetP, etc. | Custom. Normally, NCBI BLAST DBs, InterPro and UniProt-GOA | Based on Nextflow pipeline framework and software containers. |
| GenSAS [15] | Web service | BLAST+, DIAMOND, Interproscan, SignalP, TargetP, etc. | SwissProt/TrEMBL, RefSeq, RepBase | No installation needed. Requires web user registration. Includes gene structural annotation and visualization. There can be resources and usage restrictions. |
| MicrobeAnnotator [18] | Local installation | BLAST+, DIAMOND, KOFAM. | SwissProt/TrEMBL, RefSeq, KEGG | Focused on microbiomes. Conda/Python based. |
| PANNZER2 [17] | Web service | SANSparallel | UniProt, UniProt-GOA, GO, KEGG | Available command-line tool for querying the service. |
| Sma3s [19] | Local installation | BLAST+ | Reference datasets generated from UniProt, GO | A Perl script. Simple installation. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlasova, A.; Hermoso Pulido, T.; Camara, F.; Ponomarenko, J.; Guigó, R. FA-nf: A Functional Annotation Pipeline for Proteins from Non-Model Organisms Implemented in Nextflow. Genes 2021, 12, 1645. https://doi.org/10.3390/genes12101645
Vlasova A, Hermoso Pulido T, Camara F, Ponomarenko J, Guigó R. FA-nf: A Functional Annotation Pipeline for Proteins from Non-Model Organisms Implemented in Nextflow. Genes. 2021; 12(10):1645. https://doi.org/10.3390/genes12101645
Chicago/Turabian StyleVlasova, Anna, Toni Hermoso Pulido, Francisco Camara, Julia Ponomarenko, and Roderic Guigó. 2021. "FA-nf: A Functional Annotation Pipeline for Proteins from Non-Model Organisms Implemented in Nextflow" Genes 12, no. 10: 1645. https://doi.org/10.3390/genes12101645
APA StyleVlasova, A., Hermoso Pulido, T., Camara, F., Ponomarenko, J., & Guigó, R. (2021). FA-nf: A Functional Annotation Pipeline for Proteins from Non-Model Organisms Implemented in Nextflow. Genes, 12(10), 1645. https://doi.org/10.3390/genes12101645