
Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility


Abstract
1. Introduction
2. Materials and Methods
2.1. Studied Population
2.2. Gene Panel Design
- (i)
- Infertility genes: Genes defined by Online Mendelian Inheritance in Man (OMIM), at the time of design, as responsible for a non-syndromic male and/or female infertility phenotype; coded as spermatogenic failure (SPGF) for male infertility, and as premature ovarian failure (POF) and as oocyte maturation defect (OOMD) for female infertility.
- (ii)
- Candidate genes: Genes for which at least one variant potentially pathogenic for the related phenotype in humans has been identified by good-quality WES studies, but which need further confirmation.
- (iii)
- FMR1 sequencing: There is an association between pre-mutation of the FMR1 gene and increased susceptibility to idiopathic POI. We added FMR1 on the gene list in order to elucidate possible disease-causing variants for POI.
2.3. Gene Panel Sequencing
2.4. Data Analysis
2.5. Identity Control and Confirmation of Mutations
3. Results
3.1. Cohort Description
3.2. Validation of the Infertility Panel and Identity Control
3.3. Sequencing Results and Identification of Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobs, P.A.; Strong, J.A. A Case of Human Intersexuality Having a Possible XXY Sex-Determining Mechanism. Nature 1959, 183, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Tiepolo, L.; Zuffardi, O. Localization of Factors Controlling Spermatogenesis in the Nonfluorescent Portion of the Human Y Chromosome Long Arm. Hum. Genet. 1976, 34, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Reijo, R.; Lee, T.Y.; Salo, P.; Alagappan, R.; Brown, L.G.; Rosenberg, M.; Rozen, S.; Jaffe, T.; Straus, D.; Hovatta, O. Diverse Spermatogenic Defects in Humans Caused by Y Chromosome Deletions Encompassing a Novel RNA-Binding Protein Gene. Nat. Genet. 1995, 10, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.H.; Edelmann, A.; Kirsch, S.; Henegariu, O.; Hirschmann, P.; Kiesewetter, F.; Köhn, F.M.; Schill, W.B.; Farah, S.; Ramos, C.; et al. Human Y Chromosome Azoospermia Factors (AZF) Mapped to Different Subregions in Yq11. Hum. Mol. Genet. 1996, 5, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Oud, M.S.; Volozonoka, L.; Smits, R.M.; Vissers, L.E.L.M.; Ramos, L.; Veltman, J.A. A Systematic Review and Standardized Clinical Validity Assessment of Male Infertility Genes. Hum. Reprod. 2019, 34, 932–941. [Google Scholar] [CrossRef]
- França, M.M.; Funari, M.F.A.; Nishi, M.Y.; Narcizo, A.M.; Domenice, S.; Costa, E.M.F.; Lerario, A.M.; Mendonca, B.B. Identification of the First Homozygous 1-Bp Deletion in GDF9 Gene Leading to Primary Ovarian Insufficiency by Using Targeted Massively Parallel Sequencing. Clin. Genet. 2018, 93, 408–411. [Google Scholar] [CrossRef]
- Riera-Escamilla, A.; Enguita-Marruedo, A.; Moreno-Mendoza, D.; Chianese, C.; Sleddens-Linkels, E.; Contini, E.; Benelli, M.; Natali, A.; Colpi, G.M.; Ruiz-Castañé, E.; et al. Sequencing of a “mouse Azoospermia” Gene Panel in Azoospermic Men: Identification of RNF212 and STAG3 Mutations as Novel Genetic Causes of Meiotic Arrest. Hum. Reprod. 2019, 34, 978–988. [Google Scholar] [CrossRef]
- Lorenzi, D.; Fernández, C.; Bilinski, M.; Fabbro, M.; Galain, M.; Menazzi, S.; Miguens, M.; Perassi, P.N.; Fulco, M.F.; Kopelman, S.; et al. First Custom Next-Generation Sequencing Infertility Panel in Latin America: Design and First Results. JBRA Assist. Reprod. 2020, 24, 104–114. [Google Scholar] [CrossRef]
- Rocca, M.S.; Msaki, A.; Ghezzi, M.; Cosci, I.; Pilichou, K.; Celeghin, R.; Foresta, C.; Ferlin, A. Development of a Novel Next-Generation Sequencing Panel for Diagnosis of Quantitative Spermatogenic Impairment. J. Assist. Reprod. Genet. 2020, 37, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Cannarella, R.; Condorelli, R.A.; Paolacci, S.; Barbagallo, F.; Guerri, G.; Bertelli, M.; La Vignera, S.; Calogero, A.E. Next-Generation Sequencing: Toward an Increase in the Diagnostic Yield in Patients with Apparently Idiopathic Spermatogenic Failure. Asian J. Androl. 2021, 23, 24–29. [Google Scholar] [CrossRef]
- Cannarella, R.; Precone, V.; Guerri, G.; Busetto, G.M.; Di Renzo, G.C.; Gerli, S.; Manara, E.; Dautaj, A.; Bertelli, M.; Calogero, A.E. Clinical Evaluation of a Custom Gene Panel as a Tool for Precision Male Infertility Diagnosis by Next-Generation Sequencing. Life 2020, 10, 242. [Google Scholar] [CrossRef]
- BB-0033-00081; Contract ref. 17 008 C, Request no 20161013; CRB Germethèque: Toulouse, France, 2016.
- Okutman, O.; Rhouma, M.B.; Benkhalifa, M.; Muller, J.; Viville, S. Genetic Evaluation of Patients with Non-Syndromic Male Infertility. J. Assist. Reprod. Genet. 2018, 35, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Redin, C.; Gérard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient Strategy for the Molecular Diagnosis of Intellectual Disability Using Targeted High-Throughput Sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Montaut, S.; Tranchant, C.; Drouot, N.; Rudolf, G.; Guissart, C.; Tarabeux, J.; Stemmelen, T.; Velt, A.; Fourrage, C.; Nitschké, P.; et al. Assessment of a Targeted Gene Panel for Identification of Genes Associated With Movement Disorders. JAMA Neurol. 2018, 75, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Rey, T.; Tarabeux, J.; Gerard, B.; Delbarre, M.; Le Béchec, A.; Stoetzel, C.; Prasad, M.; Laugel-Haushalter, V.; Kawczynski, M.; Muller, J.; et al. Protocol GenoDENT: Implementation of a New NGS Panel for Molecular Diagnosis of Genetic Disorders with Orodental Involvement. Methods Mol. Biol. 2019, 1922, 407–452. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Geoffroy, V.; Pizot, C.; Redin, C.; Piton, A.; Vasli, N.; Stoetzel, C.; Blavier, A.; Laporte, J.; Muller, J. VaRank: A Simple and Powerful Tool for Ranking Genetic Variants. PeerJ 2015, 3, e796. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Okutman, O.; Muller, J.; Baert, Y.; Serdarogullari, M.; Gultomruk, M.; Piton, A.; Rombaut, C.; Benkhalifa, M.; Teletin, M.; Skory, V.; et al. Exome Sequencing Reveals a Nonsense Mutation in TEX15 Causing Spermatogenic Failure in a Turkish Family. Hum. Mol. Genet. 2015, 24, 5581–5588. [Google Scholar] [CrossRef] [PubMed]
- Backenroth, D.; Homsy, J.; Murillo, L.R.; Glessner, J.; Lin, E.; Brueckner, M.; Lifton, R.; Goldmuntz, E.; Chung, W.K.; Shen, Y. CANOES: Detecting Rare Copy Number Variants from Whole Exome Sequencing Data. Nucleic Acids Res. 2014, 42, e97. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An Integrated Tool for Structural Variations Annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP Sequence Variant Classification Guidelines for Single-Gene Copy Number Variants. Genet. Med. 2020, 22, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Okutman, O.; Muller, J.; Skory, V.; Garnier, J.M.; Gaucherot, A.; Baert, Y.; Lamour, V.; Serdarogullari, M.; Gultomruk, M.; Röpke, A.; et al. A No-Stop Mutation in MAGEB4 Is a Possible Cause of Rare X-Linked Azoospermia and Oligozoospermia in a Consanguineous Turkish Family. J. Assist. Reprod. Genet. 2017, 34, 683–694. [Google Scholar] [CrossRef]
- Koscinski, I.; Elinati, E.; Fossard, C.; Redin, C.; Muller, J.; de la Calle, J.V.; Schmitt, F.; Ben Khelifa, M.; Ray, P.F.; Ray, P.; et al. DPY19L2 Deletion as a Major Cause of Globozoospermia. Am. J. Hum. Genet. 2011, 88, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Araujo, T.F.; Friedrich, C.; Grangeiro, C.H.P.; Martelli, L.R.; Grzesiuk, J.D.; Emich, J.; Wyrwoll, M.J.; Kliesch, S.; Simões, A.L.; Tüttelmann, F. Sequence Analysis of 37 Candidate Genes for Male Infertility: Challenges in Variant Assessment and Validating Genes. Andrology 2020, 8, 434–441. [Google Scholar] [CrossRef]
- Dieterich, K.; Zouari, R.; Harbuz, R.; Vialard, F.; Martinez, D.; Bellayou, H.; Prisant, N.; Zoghmar, A.; Guichaoua, M.R.; Koscinski, I.; et al. The Aurora Kinase C c.144delC Mutation Causes Meiosis I Arrest in Men and Is Frequent in the North African Population. Hum. Mol. Genet. 2009, 18, 1301–1309. [Google Scholar] [CrossRef]
- Coutton, C.; Vargas, A.S.; Amiri-Yekta, A.; Kherraf, Z.-E.; Ben Mustapha, S.F.; Le Tanno, P.; Wambergue-Legrand, C.; Karaouzène, T.; Martinez, G.; Crouzy, S.; et al. Mutations in CFAP43 and CFAP44 Cause Male Infertility and Flagellum Defects in Trypanosoma and Human. Nat. Commun. 2018, 9, 686. [Google Scholar] [CrossRef]
- Maddirevula, S.; Coskun, S.; Alhassan, S.; Elnour, A.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Arold, S.T.; Alkuraya, F.S. Female Infertility Caused by Mutations in the Oocyte-Specific Translational Repressor PATL2. Am. J. Hum. Genet. 2017, 101, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for Diagnostic Next-Generation Sequencing. Eur. J. Hum. Genet. 2016, 24, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Yatsenko, A.N.; Roy, A.; Chen, R.; Ma, L.; Murthy, L.J.; Yan, W.; Lamb, D.J.; Matzuk, M.M. Non-Invasive Genetic Diagnosis of Male Infertility Using Spermatozoal RNA: KLHL10 Mutations in Oligozoospermic Patients Impair Homodimerization. Hum. Mol. Genet. 2006, 15, 3411–3419. [Google Scholar] [CrossRef]
- Ray, P.F.; Toure, A.; Metzler-Guillemain, C.; Mitchell, M.J.; Arnoult, C.; Coutton, C. Genetic Abnormalities Leading to Qualitative Defects of Sperm Morphology or Function. Clin. Genet. 2017, 91, 217–232. [Google Scholar] [CrossRef]
- Dieterich, K.; Rifo, R.S.; Faure, A.K.; Hennebicq, S.; Ben Amar, B.; Zahi, M.; Perrin, J.; Martinez, D.; Sèle, B.; Jouk, P.S.; et al. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat. Genet. 2007, 39, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.-W.; Wang, X.; Su, Z.-Y.; Mei, L.-B.; Ji, Z.-Y.; Bao, H.; Li, P. Patients with Multiple Morphological Abnormalities of the Sperm Flagella Harbouring CFAP44 or CFAP43 Mutations Have a Good Pregnancy Outcome Following Intracytoplasmic Sperm Injection. Andrologia 2019, 51, e13151. [Google Scholar] [CrossRef]
- Takasaki, N.; Tachibana, K.; Ogasawara, S.; Matsuzaki, H.; Hagiuda, J.; Ishikawa, H.; Mochida, K.; Inoue, K.; Ogonuki, N.; Ogura, A.; et al. A Heterozygous Mutation of GALNTL5 Affects Male Infertility with Impairment of Sperm Motility. Proc. Natl. Acad. Sci. USA 2014, 111, 1120–1125. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, W.; Jiang, H.; Wu, B.-L.; Primary Ovarian Insufficiency Collaboration. Mutations in HFM1 in Recessive Primary Ovarian Insufficiency. N. Engl. J. Med. 2014, 370, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Wang, C.; Cao, J.; Shen, Y.; Jiang, H.; Liu, J.; Wu, B.L.; Zhang, W.; Wu, J. Association Analysis between HFM1 Variation and Primary Ovarian Insufficiency in Chinese Women. Clin. Genet. 2016, 89, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Zhe, J.; Chen, S.; Chen, X.; Liu, Y.; Li, Y.; Zhou, X.; Zhang, J. A Novel Heterozygous Splice-Altering Mutation in HFM1 May Be a Cause of Premature Ovarian Insufficiency. J. Ovarian Res. 2019, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Kuentz, P.; Vanden Meerschaut, F.; Elinati, E.; Nasr-Esfahani, M.H.; Gurgan, T.; Iqbal, N.; Carré-Pigeon, F.; Brugnon, F.; Gitlin, S.A.; Velez de la Calle, J.; et al. Assisted Oocyte Activation Overcomes Fertilization Failure in Globozoospermic Patients Regardless of the DPY19L2 Status. Hum. Reprod. 2013, 28, 1054–1061. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Sex | Infertility Phenotype | #Patients | |
|---|---|---|---|
| Male | Teratozoopsermia | 7 | |
| Asthenozoospermia | 1 | ||
| Sperm production defect (SPD) | Azoospermia | 30 | |
| Oligozoospermia | 20 | ||
| Mixed phenotype | 21 | ||
| Female | Oocyte maturation defect (OOMD) | 4 | |
| Premature ovarian insufficiency (POI) | 11 | ||
| Sample | Phenotype | Mutated Gene | Zygosity | Defined Mutation |
|---|---|---|---|---|
| C1 | SPD | MAGEB4 | hemi | p.*347Cysext*24 (c.1041A > T) |
| C2 | SPD | TEX15 | hom | p.Tyr710* (c.2130T > G) |
| C3 | Teratozoospermia | DPY19L2 | Het gene del and point mutation | Heterozygous DPY19L2 deletion with p.Arg290His (c.869G > A) |
| C4 | Teratozoospermia | DPY19L2 | Hom | exon 5-exon 6 deletion in DPY19L2 |
| C5 | Teratozoospermia | DPY19L2 | Hom | del DPY19L2 |
| MALE INFERTILITY | Phenotype | Gene Name | OMIM # | RefSeq | IG/CG |
| Teratozoospermia | AKAP4 | 300185 | NM_003886.2 | CG | |
| AURKC | 603495 | NM_001015878.1 | IG | ||
| BRDT | 602144 | NM_001242806.2 | IG | ||
| CFAP43 | 617558 | NM_025145.6 | IG | ||
| CFAP44 | 617559 | NM_001164496.1 | IG | ||
| DNAH1 | 603332 | NM_015512.4 | IG | ||
| DPY19L2 | 613893 | NM_173812.4 | IG | ||
| MTUS1 ** | 609589 | NM_001001924.2 | CG | ||
| PICK1 | 605926 | NM_001039583.1 | CG | ||
| SEPT12 | 611562 | NM_144605.4 | IG | ||
| SPATA16 | 609856 | NM_031955.5 | IG | ||
| Asthenozoospermia | CATSPER1 | 606389 | NM_053054.3 | IG | |
| GALNTL5 | 615133 | NM_145292.3 | CG | ||
| SLC26A8 | 608480 | NM_001193476.1 | IG | ||
| SPAG17 | 616554 | NM_206996.3 | CG | ||
| Sperm production defect (SPD) | CCDC39 | 613798 | NM_181426.1 | CG * | |
| DNAH6 | 603336 | NM_001370.1 | CG | ||
| HIWI (PIWIL1) | 605571 | NM_004764.4 | CG | ||
| HSF2 | 140581 | NM_004506.3 | CG | ||
| KLHL10 | 608778 | NM_152467.4 | IG | ||
| MAGEB4 | 300153 | NM_002367.3 | CG | ||
| MEIOB | 617670 | NM_001163560.2 | IG | ||
| NANOS1 | 608226 | NM_199461.3 | IG | ||
| NPAS2 | 603347 | NM_002518.3 | IG | ||
| NROB1 (DAX1) | 300473 | NM_000475.4 | CG * | ||
| SOHLH1 | 610224 | NM_001012415.2 | IG | ||
| SPINK2 | 605753 | NM_001271722.1 | IG | ||
| TAF4B | 601689 | NM_001293725.1 | IG | ||
| TEX11 | 300311 | NM_001003811.1 | IG | ||
| TEX14 | 605792 | NM_001201457.1 | IG | ||
| TEX15 | 605795 | NM_031271.3 | IG | ||
| Wt1 | 607102 | NM_024426.3 | CG * | ||
| ZMYND15 | 614312 | NM_001267822.1 | IG | ||
| Total fertilization problem | PLCZ1 | 608075 | NM_033123.3 | IG | |
| Phenotype | Gene Name | OMIM | RefSeq | IG/CG | |
| FEMALE INFERTILITY | Primary Ovarian Insufficiency (POI) | BMP15 | 300247 | NM_005448.2 | IG |
| FIGLA | 608697 | NM_001004311.3 | IG | ||
| FMR1 | 309550 | NM_002024.5 | S | ||
| FSHR | 136435 | NM_000145.3 | CG | ||
| GDF9 | 601918 | NM_005260.5 | IG | ||
| HFM1 | 615684 | NM_001017975.4 | IG | ||
| MCM8 | 608187 | NM_001281521.1 | IG | ||
| MCM9 | 610098 | NM_017696.2 | CG | ||
| MSH4 | 602105 | NM_002440.3 | CG | ||
| NANOS3 | 608229 | NM_001098622.2 | CG | ||
| NOBOX | 610934 | NM_001080413.3 | IG | ||
| PGRMC1 | 300435 | NM_006667.4 | CG | ||
| STAG3 | 608489 | NM_001282717.1 | IG | ||
| Oocyte Maturation Defect (OOMD) | TUBB8 | 616768 | NM_177987.2 | IG | |
| PATL2 | 614661 | NM_001145112.1 | IG | ||
| Gene Name | OMIM | RefSeq | IG/CG | ||
| Male/Female infertility (SPD/POI) | NR5A1 | 184757 | NM_004959.4 | IG | |
| SYCE1 | 611486 | NM_001143764.1 | IG | ||
| Sex | Patient Code | Phenotype | Gene Name (Refseq Id) | Coding Effect | Zygosity | Consanguinity | cNomen | pNomen | Allele Frequency (gnomAD) | ART Option |
|---|---|---|---|---|---|---|---|---|---|---|
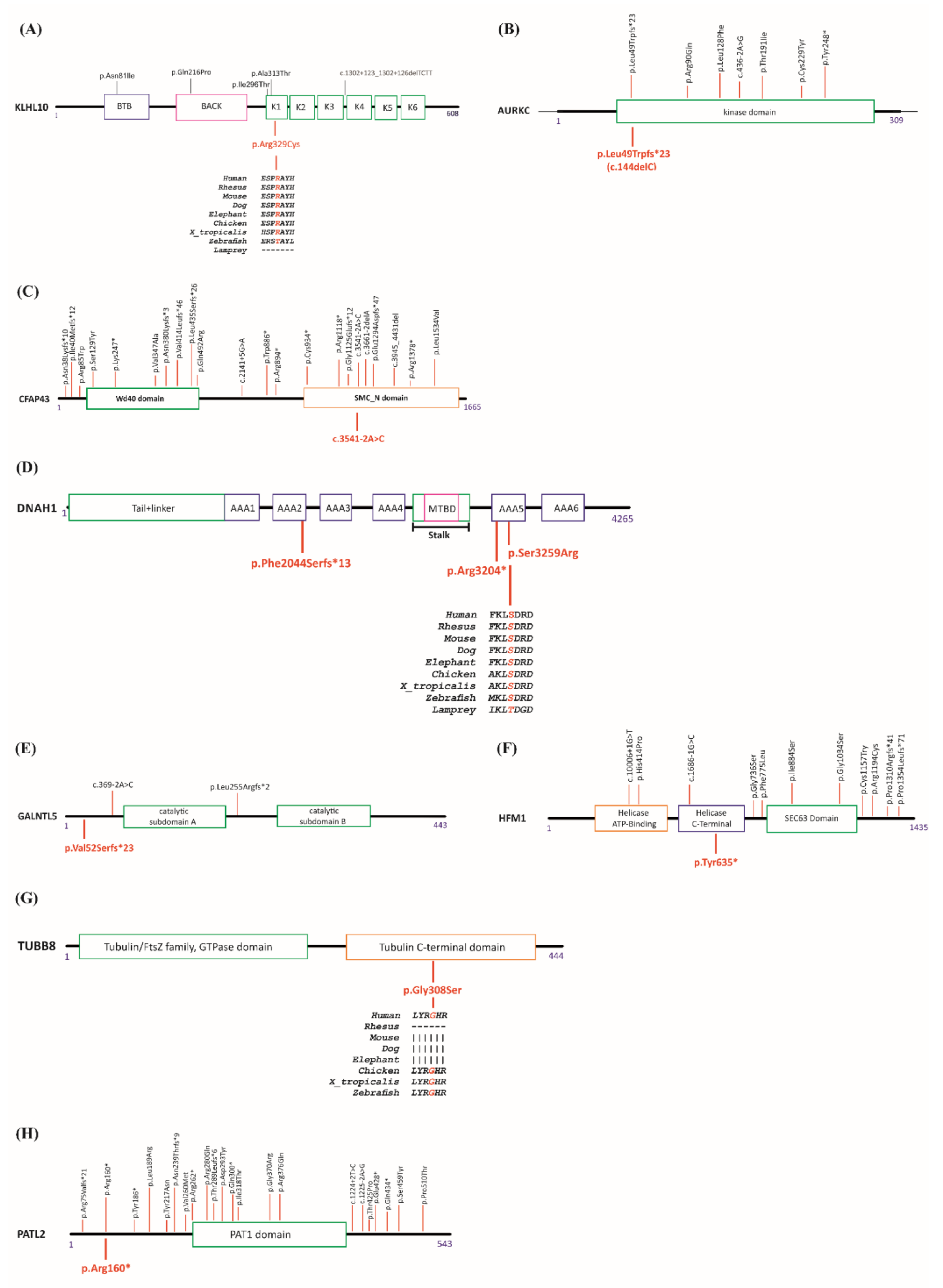
| M | Pt12 | SPD (Azoospermia) | KLHL10 (NM_001329595.1) | Missense | Het | NP | c.985C > T | p.Arg329Cys | 0.00001202 | Cryo- preservation * |
| Pt41 | Teratozoopermia | AURKC (NM_001015878) | Frameshift | Hom | No | c.144delC | p.Leu49TrpfsTer23 | 0.00008749 | Sperm donation | |
| Pt55 | Teratozoopermia (MMAF) | CFAP43 (NM_025145.5) | Splice site | Hom | NP | c.3541-2A > C | p.? | Not listed | ICSI | |
| Pt65 | AT | DNAH1 (NM_015512.4) | Stop-gain Frameshift Missense | Comp. Het | No | c.9610C > T c.6131del c.9777T > G | p.Arg3204 * p.Phe2044Serfs *13 p.Ser3259Arg | Not listed Not listed 0.000008037 | ICSI | |
| Pt77 | SPD (OAT) | GALNTL5 (NM_145292.3) | Frameshift | Het | NP | c.153dup | p.Val52Serfs*23 | Not listed | * | |
| F | Pt2 | POI | HFM1 (NM_001017975.4) | Stop-gain | Hom | Yes | c.1905T > A | p.Tyr635* | Not listed | Oocyte Donation ** |
| Pt38 | OOMD | TUBB8 (NM_177987.2) | Missense | Hom | Yes | c.922G > A | p.Gly308Ser | Not listed | Oocyte donation | |
| Pt71 | OOMD | PATL2 (NM_001145112.1) | Stop-gain | Hom | Yes | c.478C > T | p.Arg160* | 0.00003245 | Oocyte donation |
| Infertility Type | #Genes | #Cases (Male/ Female- Phenotype) | HTS Quality | Filtered-Out Frequency | Diagnostic Yield | References | ||
|---|---|---|---|---|---|---|---|---|
| Mean Coverage | Depth of Coverage | % Targetted Bases | ||||||
| Syndromic/ non-syndromic | 284 | 48 idiopathic POI females | 145X | 10X | 99.38% | > 0.1% | 2% | [6] |
| Male infertility (genes based on mouse model) | 175 | 33 idiopathic NOA | 300X | NP | NP | > 5% | 6.3% | [7] |
| Syndromic/ non-syndromic | 75 | 17 female, 6 male with different infertility phenotype | 180 | 20X | 98% | > 5% | 8.7% | [8] |
| Syndromic/ non-syndromic | 9 | 241 idiopathic male infertility cases | 351X | 10X | 93.5% | > 1% | 0.4% | [9] |
| Syndromic/ non-syndromic | 15 | 25 idiopathic male infertility with SPD | ND | ND | ND | ND | 12% | [10] |
| Syndromic/ non-syndromic | 110 | 22 male infertility cases | 286–539X * | 10X * | 91.3–98% * | ND | 25% | [11] |
| Non-syndromic | 51 | 15 female, 79 male with different infertility phenotype | 457X | 30X | 99.8% | >1% | 8.5% | Present study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okutman, O.; Tarabeux, J.; Muller, J.; Viville, S. Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility. Genes 2021, 12, 410. https://doi.org/10.3390/genes12030410
Okutman O, Tarabeux J, Muller J, Viville S. Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility. Genes. 2021; 12(3):410. https://doi.org/10.3390/genes12030410
Chicago/Turabian StyleOkutman, Ozlem, Julien Tarabeux, Jean Muller, and Stéphane Viville. 2021. "Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility" Genes 12, no. 3: 410. https://doi.org/10.3390/genes12030410
APA StyleOkutman, O., Tarabeux, J., Muller, J., & Viville, S. (2021). Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility. Genes, 12(3), 410. https://doi.org/10.3390/genes12030410