
Clustering Single-Cell RNA-Seq Data with Regularized Gaussian Graphical Model


Abstract
1. Introduction
2. Methods
2.1. -Based Regularized Gaussian Graphical Model
2.2. The Grouping Effects of -Based Regularization
2.3. Determination and Performance Measures
3. Results
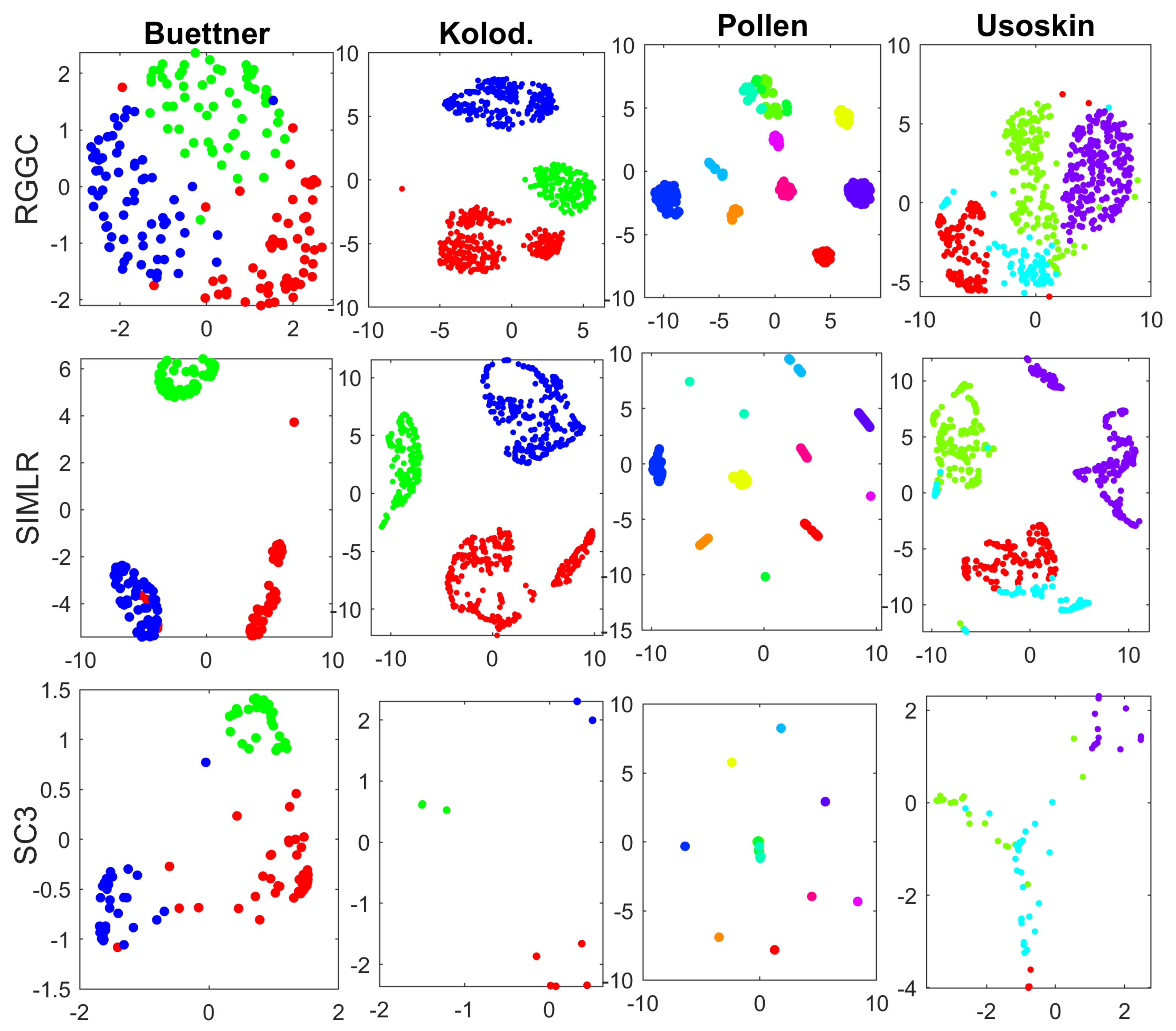
3.1. Benchmark and Simulation Data
3.2. Performance Evaluation
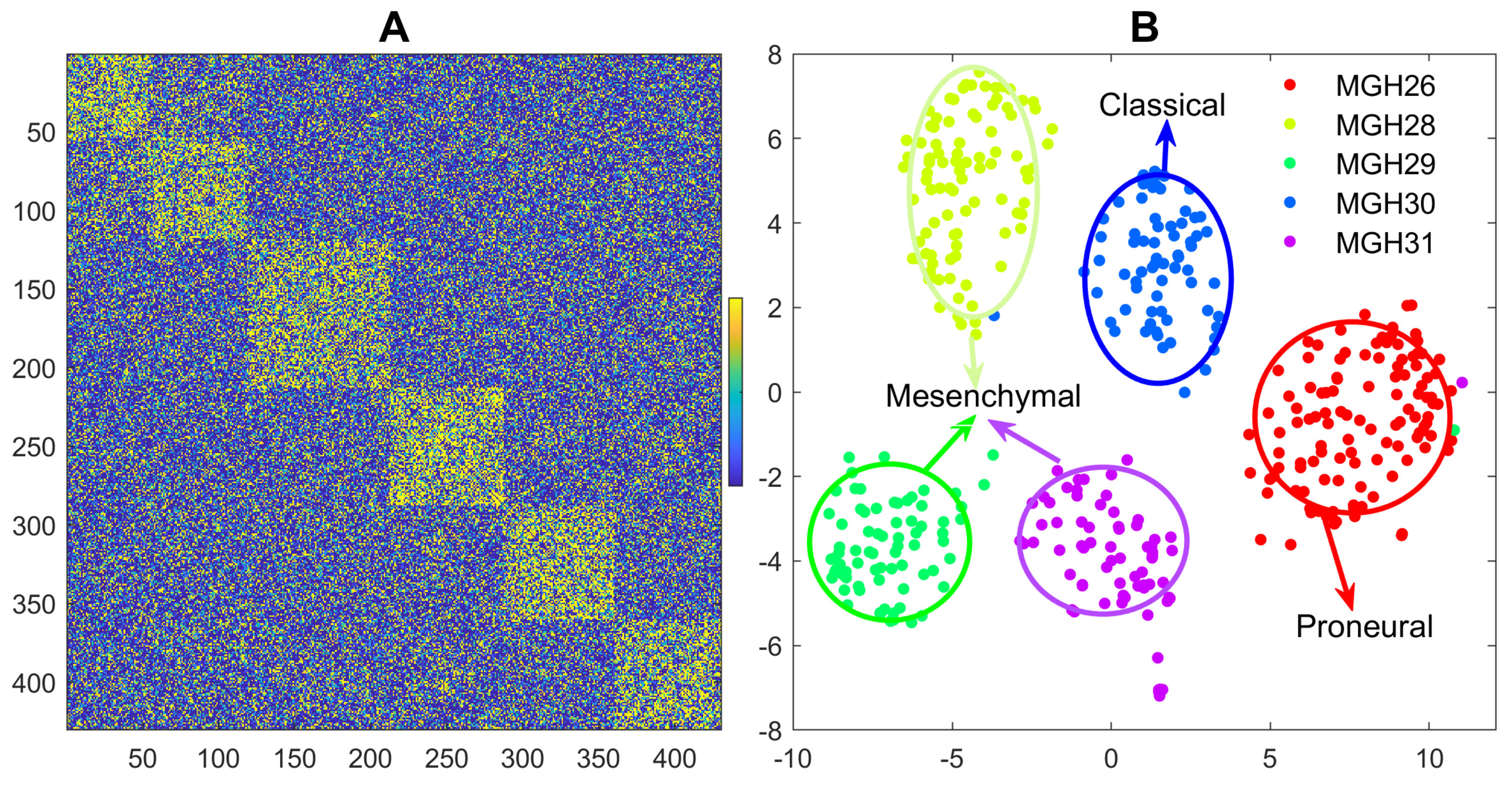
3.3. Reanalyzing the Glioblastoma scRNA-seq Data
4. Discussion and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.; Burdziak, C.; Moon, K.; Chaffer, C.; Pattabiraman, D.; et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 2018, 174, 716–729.e27. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, J.; Pierson, E.; Ramazzotti, D.; Batzoglou, S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods 2017, 14, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling Leffler, J.; Haeggström, J.; Kharchenko, O.; Kharchenko, P.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2014, 18, 145–153. [Google Scholar] [CrossRef]
- Sandberg, R. Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods 2014, 11, 22–24. [Google Scholar] [CrossRef]
- Xu, D.; Tian, Y. A Comprehensive Survey of Clustering Algorithms. Ann. Data Sci. 2015, 2, 165–193. [Google Scholar] [CrossRef]
- Yang, L.; Liu, J.; Lu, Q.; Riggs, A.; Wu, X. SAIC: An iterative clustering approach for analysis of single cell RNA-seq data. BMC Genom. 2017, 18, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef]
- Grün, D.; Muraro, M.; Boisset, J.C.; Wiebrands, K.; Lyubimova, A.; Dharmadhikari, G.; van den Born, M.; Es, J.; Jansen, E.; Clevers, H.; et al. De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data. Cell Stem Cell 2016, 19, 266–277. [Google Scholar] [CrossRef]
- Kiselev, V.; Kirschner, K.; Schaub, M.; Andrews, T.; Yiu, A.; Chandra, T.; Natarajan, K.; Reik, W.; Barahona, M.; Green, A.; et al. SC3: Consensus clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2017, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Žurauskienė, J.; Yau, C. pcaReduce: Hierarchical clustering of single cell transcriptional profiles. BMC Bioinform. 2016, 17, 140. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Levine, J.; Simonds, E.; Bendall, S.; Davis, K.; Amir, E.A.; Tadmor, M.; Litvin, O.; Fienberg, H.; Jager, A.; Zunder, E.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef]
- Macosko, E.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.; Kamitaki, N.; Martersteck, E.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.; Angerer, P.; Theis, F. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Fortunato, S.; Barthelemy, M. Resolution limit in community detection. Proc. Natl. Acad. Sci. USA 2007, 104, 36–41. [Google Scholar] [CrossRef]
- Ding, J.; Condon, A.; Shah, S. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 2018, 9, 2002. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gu, J. VASC: Dimension Reduction and Visualization of Single-cell RNA-seq Data by Deep Variational Autoencoder. Genom. Proteom. Bioinform. 2018, 16, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, X.; Lin, Q.; Wong, K.C. Review of Single-cell RNA-seq Data Clustering for Cell Type Identification and Characterization. arXiv 2020, arXiv:2001.01006. [Google Scholar]
- Kiselev, V.; Andrews, T.; Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019, 20, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Rupp, M.; Schneider, P.; Schneider, G. Distance phenomena in high-dimensional chemical descriptor spaces: Consequences for similarity-based approaches. J. Comput. Chem. 2009, 30, 2285–2296. [Google Scholar]
- Pourkamali-Anaraki, F.; Becker, S. Efficient Solvers for Sparse Subspace Clustering. Signal Process. 2018, 172, 107548. [Google Scholar] [CrossRef]
- Liu, Z.; Lin, S.; Piantadosi, S. Network construction and structure detection with metagenomic count data. BioData Min. 2015, 8, 40. [Google Scholar] [CrossRef][Green Version]
- Liu, Z.; Sun, F.; Braun, J.; McGovern, D.; Piantadosi, S. Multilevel regularized regression for simultaneous taxa selection and network construction with metagenomic count data. Bioinformatics 2015, 31, 1067–1074. [Google Scholar] [CrossRef]
- Canyi, L.; Hai, M.; Zhao, Z.Q.; Zhu, L.; Huang, D.S.; Yan, S. Robust and Efficient Subspace Segmentation via Least Squares Regression. In Proceedings of the 12th European conference on Computer Vision—Volume Part VII, Florence, Italy, 7–13 October 2012; Volume 7578, pp. 347–360. [Google Scholar] [CrossRef]
- Vinh, N.; Epps, J.; Bailey, J. Information Theoretic Measures for Clusterings Comparison: Variants, Properties, Normalization and Correction for Chance. J. Mach. Learn. Res. 2010, 11, 2837–2854. [Google Scholar]
- Van der Maaten, L. Accelerating t-SNE using Tree-Based Algorithms. J. Mach. Learn. Res. 2015, 15, 3221–3245. [Google Scholar]
- Buettner, F.; Natarajan, K.; Casale, F.; Proserpio, V.; Scialdone, A.; Theis, F.; Teichmann, S.; Marioni, J.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-Sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Tsang, J.; Ilicic, T.; Henriksson, J.; Natarajan, K.; Tuck, A.; Gao, X.; Bühler, M.; Liu, P.; et al. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell 2015, 17, 471–485. [Google Scholar] [CrossRef]
- Pollen, A.; Nowakowski, T.; Shuga, J.; Wang, X.; Leyrat, A.; Lui, J.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 2014, 32, 1053. [Google Scholar] [CrossRef] [PubMed]
- Zappia, L.; Phipson, B.; Oshlack, A. Splatter: Simulation of single-cell RNA sequencing data. Genome Biol. 2017, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Li, M.; Liang, Z.; Wu, F.X.; Pan, Y.; Wang, J. SinNLRR: A robust subspace clustering method for cell type detection by nonnegative and low rank representation. Bioinformatics 2019, 35, 3642–3650. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z. Visualizing Single-Cell RNA-seq Data with Semisupervised Principal Component Analysis. Int. J. Mol. Sci. 2020, 21, 5797. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Tirosh, I.; Trombetta, J.; Shalek, A.; Gillespie, S.; Wakimoto, H.; Cahill, D.; Nahed, B.; Curry, W.T.; Martuza, R.; et al. Single-cell RNA-Seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.; Hoadley, K.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| NMI | ARI | |||||||
|---|---|---|---|---|---|---|---|---|
| Buettner | Kolod. | Pollen | Usoskin | Buettner | Kolod. | Pollen | Usoskin | |
| 0.00 | 0.88 | 0.84 | 0.93 | 0.96 | 0.92 | 0.81 | 0.91 | 0.98 |
| 0.01 | 0.88 | 0.84 | 0.93 | 0.90 | 0.92 | 0.81 | 0.91 | 0.93 |
| 0.1 | 0.88 | 0.84 | 0.93 | 0.96 | 0.92 | 0.81 | 0.91 | 0.98 |
| 1.0 | 0.88 | 0.90 | 0.93 | 0.96 | 0.92 | 0.81 | 0.91 | 0.98 |
| 2.0 (AIC) | 0.90 | 0.84 | 0.93 | 0.96 | 0.92 | 0.81 | 0.91 | 0.98 |
| logp (BIC) | 0.88 | 0.84 | 0.93 | 0.96 | 0.92 | 0.81 | 0.91 | 0.98 |
| 2logp | 0.88 | 0.84 | 0.93 | 0.96 | 0.92 | 0.81 | 0.91 | 0.98 |
| 100 | 0.88 | 0.84 | 0.93 | 0.96 | 0.92 | 0.81 | 0.91 | 0.98 |
| 1000 | 0.88 | 0.83 | 0.90 | 0.96 | 0.92 | 0.79 | 0.87 | 0.98 |
| Methods | NMI | ARI | ||||
|---|---|---|---|---|---|---|
| SimData I | SimData II | SimData III | SimData I | SimData II | SimData III | |
| PCA | 0.67 | 0.70 | 0.72 | 0.65 | 0.70 | 0.68 |
| RGGC | 0.96 | 0.97 | 0.98 | 0.97 | 0.99 | 0.99 |
| SIMLR | 0.74 | 0.89 | 0.85 | 0.71 | 0.90 | 0.87 |
| SC3 | 0.94 | 93 | 0.96 | 0.95 | 0.95 | 0.97 |
| RaceID2 | 0.97 | 0.97 | 0.76 | 0.98 | 0.98 | 0.63 |
| Seurat | 0.86 | 0.90 | 0.94 | 0.71 | 0.86 | 0.93 |
| SSC(L1) | 0.72 | 0.61 | 0.71 | 0.64 | 0.57 | 0.57 |
| SinNLRR | 0.73 | 0.78 | 0.83 | 0.66 | 0.79 | 0.81 |
| Methods | NMI | ARI | ||||||
|---|---|---|---|---|---|---|---|---|
| Buettner | Kolod. | Pollen | Usoskin | Buettner | Kolod. | Pollen | Usoskin | |
| PCA | 0.56 | 0.77 | 0.83 | 0.39 | 0.59 | 0.75 | 0.77 | 0.44 |
| RGGC | 0.88 | 0.84 | 0.93 | 0.96 | 0.92 | 0.81 | 0.91 | 0.98 |
| SIMLR | 0.91 | 0.99 | 0.95 | 0.74 | 0.92 | 0.99 | 0.94 | 0.68 |
| SC3 | 0.83 | 1.0 | 0.96 | 0.87 | 0.86 | 1.0 | 0.95 | 0.88 |
| RaceID2 | 0.31 | 0.66 | 0.92 | 0.35 | 0.32 | 0.62 | 0.91 | 0.34 |
| Seurat | 0.56 | 0.92 | 0.90 | 0.74 | 0.39 | 0.90 | 0.75 | 0.57 |
| SSC(L1) | 0.72 | 0.64 | 0.91 | 0.61 | 0.78 | 0.61 | 0.86 | 0.55 |
| SinNLRR | 0.60 | 0.78 | 0.93 | 0.85 | 0.63 | 0.72 | 0.91 | 0.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z. Clustering Single-Cell RNA-Seq Data with Regularized Gaussian Graphical Model. Genes 2021, 12, 311. https://doi.org/10.3390/genes12020311
Liu Z. Clustering Single-Cell RNA-Seq Data with Regularized Gaussian Graphical Model. Genes. 2021; 12(2):311. https://doi.org/10.3390/genes12020311
Chicago/Turabian StyleLiu, Zhenqiu. 2021. "Clustering Single-Cell RNA-Seq Data with Regularized Gaussian Graphical Model" Genes 12, no. 2: 311. https://doi.org/10.3390/genes12020311
APA StyleLiu, Z. (2021). Clustering Single-Cell RNA-Seq Data with Regularized Gaussian Graphical Model. Genes, 12(2), 311. https://doi.org/10.3390/genes12020311