
The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases

 , , , ,
, , , ,
Abstract
1. Introduction
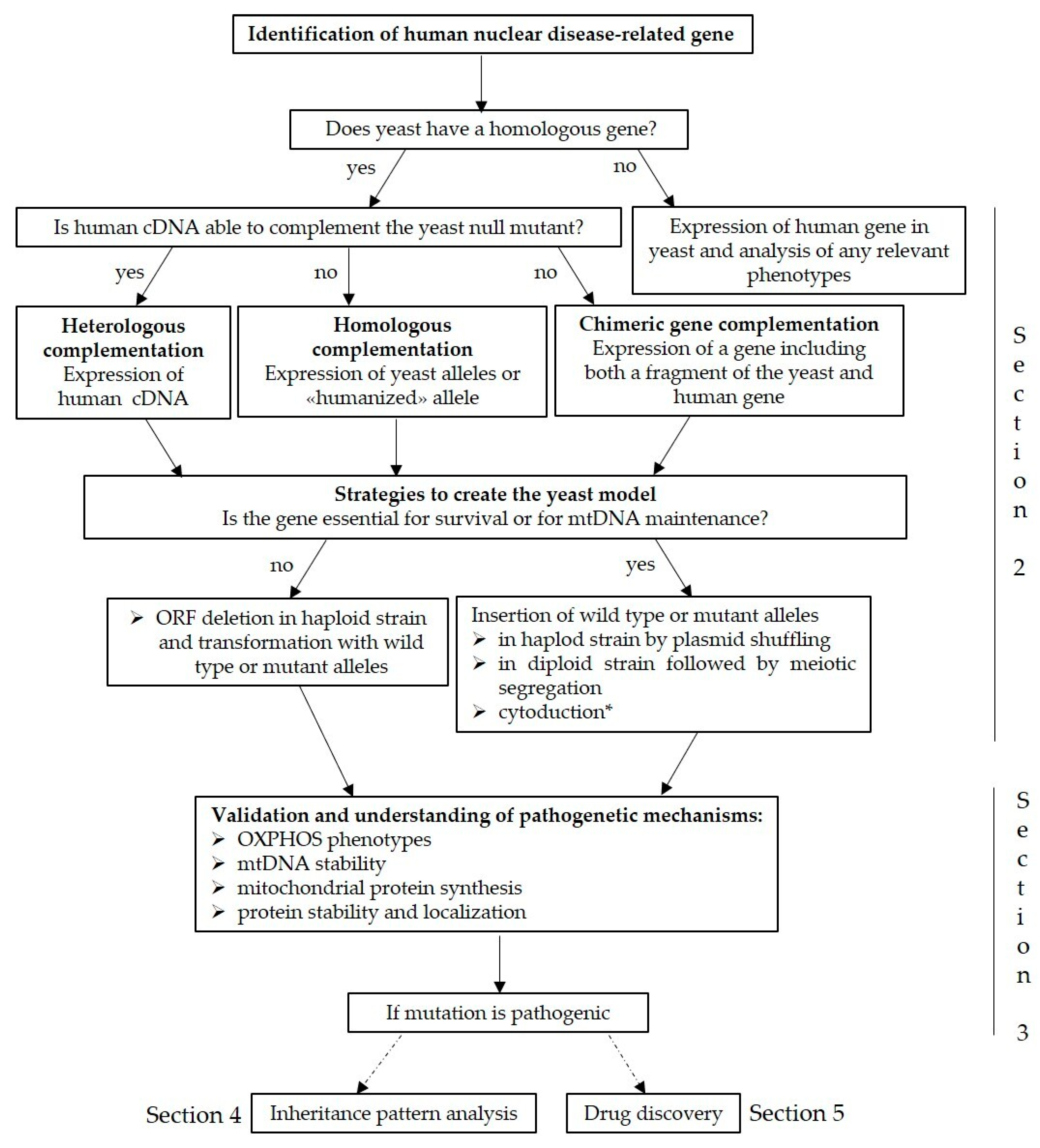
2. Modelling Putative Disease Variants
3. Validation of Mutations and Understanding of Pathogenetic Mechanisms
3.1. Analysis of OXPHOS Phenotypes
3.2. Determination of mtDNA Stability
3.3. Analysis of Mitochondrial Protein Synthesis (MPS)
3.4. Evaluation of Protein Stability
3.5. Analysis of Mutant Proteins Localisation
4. Inheritance Pattern Analysis: Dominance/Recessivity and Gene Interactions Analysis
5. Yeast as A Model for Mitochondrial Diseases Drug Discovery
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Oláhová, M.; McFarland, R.; Taylor, R.W. Recent Advances in Understanding the Molecular Genetic Basis of Mitochondrial Disease. J. Inherit. Metab. Dis. 2020, 43, 36–50. [Google Scholar] [CrossRef]
- Zeviani, M.; Di Donato, S. Mitochondrial Disorders. Brain 2004, 127, 2153–2172. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P.; Bourgeron, T.; Parfait, B.; Chretien, D.; Munnich, A.; Rötig, A. Inborn Errors of the Krebs Cycle: A Group of Unusual Mitochondrial Diseases in Human. Biochim. Biophys. Acta 1997, 1361, 185–197. [Google Scholar] [CrossRef]
- Hiltunen, J.K.; Schonauer, M.S.; Autio, K.J.; Mittelmeier, T.M.; Kastaniotis, A.J.; Dieckmann, C.L. Mitochondrial Fatty Acid Synthesis Type II: More than Just Fatty Acids. J. Biol. Chem. 2009, 284, 9011–9015. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional Mitochondria in Health and Disease. Front. Endocrinol. 2017, 8, 296. [Google Scholar] [CrossRef]
- Rawat, S.; Stemmler, T.L. Key Players and Their Role during Mitochondrial Iron-Sulfur Cluster Biosynthesis. Chemistry 2011, 17, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.G. Diagnosing and Exploiting Cancer’s Addiction to Blocks in Apoptosis. Nat. Rev. Cancer 2008, 8, 121–132. [Google Scholar] [CrossRef]
- Rimessi, A.; Giorgi, C.; Pinton, P.; Rizzuto, R. The Versatility of Mitochondrial Calcium Signals: From Stimulation of Cell Metabolism to Induction of Cell Death. Biochim. Biophys. Acta 2008, 1777, 808–816. [Google Scholar] [CrossRef]
- Dimauro, S.; Davidzon, G. Mitochondrial DNA and Disease. Ann. Med. 2005, 37, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Carelli, V. Mitochondrial Disorders. Curr. Opin. Neurol. 2007, 20, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Isohanni, P. Mitochondrial DNA Depletion Syndromes--Many Genes, Common Mechanisms. Neuromuscul. Disord. 2010, 20, 429–437. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial Disorders as Windows into an Ancient Organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The Epidemiology of Mitochondrial Disorders--Past, Present and Future. Biochim. Biophys. Acta 2004, 1659, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Scapagini, G.; Currò, D.; Giuffrida Stella, A.M.; De Marco, C.; Butterfield, D.A.; Calabrese, V. Mitochondrial Dysfunction, Free Radical Generation and Cellular Stress Response in Neurodegenerative Disorders. Front. Biosci. 2007, 12, 1107–1123. [Google Scholar] [CrossRef]
- Ng, Y.S.; Turnbull, D.M. Mitochondrial Disease: Genetics and Management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA Mutations in Neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411. [Google Scholar] [CrossRef]
- Ahuja, P.; Wanagat, J.; Wang, Z.; Wang, Y.; Liem, D.A.; Ping, P.; Antoshechkin, I.A.; Margulies, K.B.; Maclellan, W.R. Divergent Mitochondrial Biogenesis Responses in Human Cardiomyopathy. Circulation 2013, 127, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Prokisch, H. Advancing Genomic Approaches to the Molecular Diagnosis of Mitochondrial Disease. Essays Biochem. 2018, 62, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Lasserre, J.-P.; Dautant, A.; Aiyar, R.S.; Kucharczyk, R.; Glatigny, A.; Tribouillard-Tanvier, D.; Rytka, J.; Blondel, M.; Skoczen, N.; Reynier, P.; et al. Yeast as a System for Modeling Mitochondrial Disease Mechanisms and Discovering Therapies. Dis. Model. Mech. 2015, 8, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An Updated Inventory of Mammalian Mitochondrial Proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B. Current Progress with Mammalian Models of Mitochondrial DNA Disease. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef]
- Ephrussi, B.; Slonimski, P.P. Subcellular Units Involved in the Synthesis of Respiratory Enzymes in Yeast. Nature 1955, 176, 1207–1208. [Google Scholar] [CrossRef]
- Schatz, G.; Haslbrunner, E.; Tuppy, H. Deoxyribonucleic acid associated with yeast mitochondria. Biochem. Biophys Res. Commun. 1964, 15, 127–132. [Google Scholar] [CrossRef]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional Characterization of the S. Cerevisiae Genome by Gene Deletion and Parallel Analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef]
- Guthrie, C.; Fink, G. Guide to Yeast Genetics and Molecular Biology. In Methods in Enzymology; Academic Press: San Diego, CA, USA, 1991; Volume 194, pp. 3–933. [Google Scholar]
- Guthrie, C.; Fink, G. Guide to Yeast Genetics and Molecular and Cell Biology—Part B. In Methods in Enzymology; Academic Press: San Diego, CA, USA, 2002; Volume 350, pp. 3–623. [Google Scholar]
- Foury, F. Human Genetic Diseases: A Cross-Talk between Man and Yeast. Gene 1997, 195, 1–10. [Google Scholar] [CrossRef]
- Foury, F.; Kucej, M. Yeast Mitochondrial Biogenesis: A Model System for Humans? Curr. Opin. Chem. Biol. 2002, 6, 106–111. [Google Scholar] [CrossRef]
- Prokisch, H.; Scharfe, C.; Camp, D.G.; Xiao, W.; David, L.; Andreoli, C.; Monroe, M.E.; Moore, R.J.; Gritsenko, M.A.; Kozany, C.; et al. Integrative Analysis of the Mitochondrial Proteome in Yeast. PLoS Biol. 2004, 2, e160. [Google Scholar] [CrossRef] [PubMed]
- Reinders, J.; Zahedi, R.P.; Pfanner, N.; Meisinger, C.; Sickmann, A. Toward the Complete Yeast Mitochondrial Proteome: Multidimensional Separation Techniques for Mitochondrial Proteomics. J. Proteome Res. 2006, 5, 1543–1554. [Google Scholar] [CrossRef]
- Barrientos, A. Yeast Models of Human Mitochondrial Diseases. IUBMB Life 2003, 55, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Dallabona, C.; Ferrero, I.; Frontali, L.; Bolotin-Fukuhara, M. Mitochondrial Diseases and the Role of the Yeast Models. FEMS Yeast Res. 2010, 10, 1006–1022. [Google Scholar] [CrossRef]
- Baile, M.G.; Claypool, S.M. The Power of Yeast to Model Diseases of the Powerhouse of the Cell. Front. Biosci. (Landmark Ed.) 2013, 18, 241–278. [Google Scholar] [CrossRef]
- Francisci, S.; Montanari, A. Mitochondrial Diseases: Yeast as a Model for the Study of Suppressors. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 666–673. [Google Scholar] [CrossRef]
- Khurana, V.; Lindquist, S. Modelling Neurodegeneration in Saccharomyces Cerevisiae: Why Cook with Baker’s Yeast? Nat. Rev. Neurosci. 2010, 11, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T.; Rustin, P.; Chretien, D.; Birch-Machin, M.; Bourgeois, M.; Viegas-Péquignot, E.; Munnich, A.; Rötig, A. Mutation of a Nuclear Succinate Dehydrogenase Gene Results in Mitochondrial Respiratory Chain Deficiency. Nat. Genet. 1995, 11, 144–149. [Google Scholar] [CrossRef]
- Alston, C.L.; Davison, J.E.; Meloni, F.; van der Westhuizen, F.H.; He, L.; Hornig-Do, H.-T.; Peet, A.C.; Gissen, P.; Goffrini, P.; Ferrero, I.; et al. Recessive Germline SDHA and SDHB Mutations Causing Leukodystrophy and Isolated Mitochondrial Complex II Deficiency. J. Med. Genet. 2012, 49, 569–577. [Google Scholar] [CrossRef]
- Nesti, C.; Meschini, M.C.; Meunier, B.; Sacchini, M.; Doccini, S.; Romano, A.; Petrillo, S.; Pezzini, I.; Seddiki, N.; Rubegni, A.; et al. Additive Effect of Nuclear and Mitochondrial Mutations in a Patient with Mitochondrial Encephalomyopathy. Hum. Mol. Genet. 2015, 24, 3248–3256. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alston, C.L.; Ceccatelli Berti, C.; Blakely, E.L.; Oláhová, M.; He, L.; McMahon, C.J.; Olpin, S.E.; Hargreaves, I.P.; Nolli, C.; McFarland, R.; et al. A Recessive Homozygous p.Asp92Gly SDHD Mutation Causes Prenatal Cardiomyopathy and a Severe Mitochondrial Complex II Deficiency. Hum. Genet. 2015, 134, 869–879. [Google Scholar] [CrossRef]
- Chang, Y.-L.; Hsieh, M.-H.; Chang, W.-W.; Wang, H.-Y.; Lin, M.-C.; Wang, C.-P.; Lou, P.-J.; Teng, S.-C. Instability of Succinate Dehydrogenase in SDHD Polymorphism Connects Reactive Oxygen Species Production to Nuclear and Mitochondrial Genomic Mutations in Yeast. Antioxid. Redox Signal. 2015, 22, 587–602. [Google Scholar] [CrossRef]
- Massa, V.; Fernandez-Vizarra, E.; Alshahwan, S.; Bakhsh, E.; Goffrini, P.; Ferrero, I.; Mereghetti, P.; D’Adamo, P.; Gasparini, P.; Zeviani, M. Severe Infantile Encephalomyopathy Caused by a Mutation in COX6B1, a Nucleus-Encoded Subunit of Cytochrome c Oxidase. Am. J. Hum. Genet. 2008, 82, 1281–1289. [Google Scholar] [CrossRef]
- Sardin, E.; Donadello, S.; di Rago, J.P.; Tetaud, E. Biochemical investigation of a human pathogenic mutation in the nuclear ATP5E gene using yeast as a model. Front. Genet. 2015, 6, 159. [Google Scholar] [CrossRef]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, Encoding a LYR Complex-II Specific Assembly Factor, Is Mutated in SDH-Defective Infantile Leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Bugiani, M.; Goffrini, P.; Carrara, F.; Farina, L.; Procopio, E.; Donati, A.; Uziel, G.; Ferrero, I.; Zeviani, M. Impaired Complex III Assembly Associated with BCS1L Gene Mutations in Isolated Mitochondrial Encephalopathy. Hum. Mol. Genet. 2007, 16, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.L.; Fehmi, J.; Czermin, B.; Goffrini, P.; Meloni, F.; Ferrero, I.; He, L.; Blakely, E.L.; McFarland, R.; Horvath, R.; et al. Long-Term Survival of Neonatal Mitochondrial Complex III Deficiency Associated with a Novel BCS1L Gene Mutation. Mol. Genet. Metab. 2010, 100, 345–348. [Google Scholar] [CrossRef] [PubMed]
- de Lonlay, P.; Valnot, I.; Barrientos, A.; Gorbatyuk, M.; Tzagoloff, A.; Taanman, J.W.; Benayoun, E.; Chrétien, D.; Kadhom, N.; Lombès, A.; et al. A Mutant Mitochondrial Respiratory Chain Assembly Protein Causes Complex III Deficiency in Patients with Tubulopathy, Encephalopathy and Liver Failure. Nat. Genet. 2001, 29, 57–60. [Google Scholar] [CrossRef]
- Meunier, B.; Fisher, N.; Ransac, S.; Mazat, J.-P.; Brasseur, G. Respiratory Complex III Dysfunction in Humans and the Use of Yeast as a Model Organism to Study Mitochondrial Myopathy and Associated Diseases. Biochim. Biophys. Acta 2013, 1827, 1346–1361. [Google Scholar] [CrossRef]
- Oláhová, M.; Ceccatelli Berti, C.; Collier, J.J.; Alston, C.L.; Jameson, E.; Jones, S.A.; Edwards, N.; He, L.; Chinnery, P.F.; Horvath, R.; et al. Molecular Genetic Investigations Identify New Clinical Phenotypes Associated with BCS1L-Related Mitochondrial Disease. Hum. Mol. Genet. 2019, 28, 3766–3776. [Google Scholar] [CrossRef]
- Invernizzi, F.; Tigano, M.; Dallabona, C.; Donnini, C.; Ferrero, I.; Cremonte, M.; Ghezzi, D.; Lamperti, C.; Zeviani, M. A Homozygous Mutation in LYRM7/MZM1L Associated with Early Onset Encephalopathy, Lactic Acidosis, and Severe Reduction of Mitochondrial Complex III Activity. Hum. Mutat. 2013, 34, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Dallabona, C.; Abbink, T.E.M.; Carrozzo, R.; Torraco, A.; Legati, A.; van Berkel, C.G.M.; Niceta, M.; Langella, T.; Verrigni, D.; Rizza, T.; et al. LYRM7 Mutations Cause a Multifocal Cavitating Leukoencephalopathy with Distinct MRI Appearance. Brain 2016, 139, 782–794. [Google Scholar] [CrossRef]
- Valnot, I.; von Kleist-Retzow, J.C.; Barrientos, A.; Gorbatyuk, M.; Taanman, J.W.; Mehaye, B.; Rustin, P.; Tzagoloff, A.; Munnich, A.; Rötig, A. A Mutation in the Human Heme A:Farnesyltransferase Gene (COX10 ) Causes Cytochrome c Oxidase Deficiency. Hum. Mol. Genet. 2000, 9, 1245–1249. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S.; Taanman, J.-W.; Rahman, S.; Meunier, B.; Sadowski, M.; Cirak, S.; Hargreaves, I.; Land, J.M.; Nanji, T.; Polke, J.M.; et al. COX10 Mutations Resulting in Complex Multisystem Mitochondrial Disease That Remains Stable into Adulthood. JAMA Neurol. 2013, 70, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, R.; Bareth, B.; Balleininger, M.; Wissel, M.; Rehling, P.; Mick, D.U. Mimicking a SURF1 Allele Reveals Uncoupling of Cytochrome c Oxidase Assembly from Translational Regulation in Yeast. Hum. Mol. Genet. 2011, 20, 2379–2393. [Google Scholar] [CrossRef]
- Bestwick, M.; Jeong, M.-Y.; Khalimonchuk, O.; Kim, H.; Winge, D.R. Analysis of Leigh Syndrome Mutations in the Yeast SURF1 Homolog Reveals a New Member of the Cytochrome Oxidase Assembly Factor Family. Mol. Cell. Biol. 2010, 30, 4480–4491. [Google Scholar] [CrossRef]
- De Meirleir, L.; Seneca, S.; Lissens, W.; De Clercq, I.; Eyskens, F.; Gerlo, E.; Smet, J.; Van Coster, R. Respiratory Chain Complex V Deficiency Due to a Mutation in the Assembly Gene ATP12. J. Med. Genet. 2004, 41, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, A.; Seneca, S.; Pribyl, T.; Smet, J.; Alderweirldt, V.; Waeytens, A.; Lissens, W.; Van Coster, R.; De Meirleir, L.; di Rago, J.-P.; et al. Defining the Pathogenesis of the Human Atp12p W94R Mutation Using a Saccharomyces cerevisiae Yeast Model. J. Biol. Chem. 2010, 285, 4099–4109. [Google Scholar] [CrossRef]
- Di Bella, D.; Lazzaro, F.; Brusco, A.; Plumari, M.; Battaglia, G.; Pastore, A.; Finardi, A.; Cagnoli, C.; Tempia, F.; Frontali, M.; et al. Mutations in the Mitochondrial Protease Gene AFG3L2 Cause Dominant Hereditary Ataxia SCA28. Nat. Genet. 2010, 42, 313–321. [Google Scholar] [CrossRef]
- Magri, S.; Fracasso, V.; Plumari, M.; Alfei, E.; Ghezzi, D.; Gellera, C.; Rusmini, P.; Poletti, A.; Di Bella, D.; Elia, A.E.; et al. Concurrent AFG3L2 and SPG7 Mutations Associated with Syndromic Parkinsonism and Optic Atrophy with Aberrant OPA1 Processing and Mitochondrial Network Fragmentation. Hum. Mutat. 2018, 39, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Adams, D.; Bonn, F.; Martinelli, P.; Cherukuri, P.F.; Teer, J.K.; Hansen, N.F.; Cruz, P.; Mullikin For The Nisc Comparative Sequencing Program, J.C.; Blakesley, R.W.; et al. Whole-Exome Sequencing Identifies Homozygous AFG3L2 Mutations in a Spastic Ataxia-Neuropathy Syndrome Linked to Mitochondrial m-AAA Proteases. PLoS Genet. 2011, 7, e1002325. [Google Scholar] [CrossRef] [PubMed]
- Bonn, F.; Pantakani, K.; Shoukier, M.; Langer, T.; Mannan, A.U. Functional Evaluation of Paraplegin Mutations by a Yeast Complementation Assay. Hum. Mutat. 2010, 31, 617–621. [Google Scholar] [CrossRef]
- Atorino, L.; Silvestri, L.; Koppen, M.; Cassina, L.; Ballabio, A.; Marconi, R.; Langer, T.; Casari, G. Loss of M-AAA Protease in Mitochondria Causes Complex I Deficiency and Increased Sensitivity to Oxidative Stress in Hereditary Spastic Paraplegia. J. Cell Biol. 2003, 163, 777–787. [Google Scholar] [CrossRef]
- Koppen, M.; Metodiev, M.D.; Casari, G.; Rugarli, E.I.; Langer, T. Variable and Tissue-Specific Subunit Composition of Mitochondrial m-AAA Protease Complexes Linked to Hereditary Spastic Paraplegia. Mol. Cell. Biol. 2007, 27, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Di Fonzo, A.; Ronchi, D.; Lodi, T.; Fassone, E.; Tigano, M.; Lamperti, C.; Corti, S.; Bordoni, A.; Fortunato, F.; Nizzardo, M.; et al. The Mitochondrial Disulfide Relay System Protein GFER Is Mutated in Autosomal-Recessive Myopathy with Cataract and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2009, 84, 594–604. [Google Scholar] [CrossRef]
- Ceh-Pavia, E.; Ang, S.K.; Spiller, M.P.; Lu, H. The Disease-Associated Mutation of the Mitochondrial Thiol Oxidase Erv1 Impairs Cofactor Binding during Its Catalytic Reaction. Biochem. J. 2014, 464, 449–459. [Google Scholar] [CrossRef]
- Eldomery, M.K.; Akdemir, Z.C.; Vögtle, F.-N.; Charng, W.-L.; Mulica, P.; Rosenfeld, J.A.; Gambin, T.; Gu, S.; Burrage, L.C.; Al Shamsi, A.; et al. MIPEP Recessive Variants Cause a Syndrome of Left Ventricular Non-Compaction, Hypotonia, and Infantile Death. Genome Med. 2016, 8, 106. [Google Scholar] [CrossRef]
- Brunetti, D.; Torsvik, J.; Dallabona, C.; Teixeira, P.; Sztromwasser, P.; Fernandez-Vizarra, E.; Cerutti, R.; Reyes, A.; Preziuso, C.; D’Amati, G.; et al. Defective PITRM1 Mitochondrial Peptidase Is Associated with Aβ Amyloidotic Neurodegeneration. EMBO Mol. Med. 2016, 8, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Langer, Y.; Aran, A.; Gulsuner, S.; Abu Libdeh, B.; Renbaum, P.; Brunetti, D.; Teixeira, P.-F.; Walsh, T.; Zeligson, S.; Ruotolo, R.; et al. Mitochondrial PITRM1 Peptidase Loss-of-Function in Childhood Cerebellar Atrophy. J. Med. Genet. 2018, 55, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Vögtle, F.-N.; Brändl, B.; Larson, A.; Pendziwiat, M.; Friederich, M.W.; White, S.M.; Basinger, A.; Kücükköse, C.; Muhle, H.; Jähn, J.A.; et al. Mutations in PMPCB Encoding the Catalytic Subunit of the Mitochondrial Presequence Protease Cause Neurodegeneration in Early Childhood. Am. J. Hum. Genet. 2018, 102, 557–573. [Google Scholar] [CrossRef]
- Shahrour, M.A.; Staretz-Chacham, O.; Dayan, D.; Stephen, J.; Weech, A.; Damseh, N.; Pri Chen, H.; Edvardson, S.; Mazaheri, S.; Saada, A.; et al. Mitochondrial Epileptic Encephalopathy, 3-Methylglutaconic Aciduria and Variable Complex V Deficiency Associated with TIMM50 Mutations. Clin. Genet. 2017, 91, 690–696. [Google Scholar] [CrossRef]
- Roesch, K.; Curran, S.P.; Tranebjaerg, L.; Koehler, C.M. Human Deafness Dystonia Syndrome Is Caused by a Defect in Assembly of the DDP1/TIMM8a-TIMM13 Complex. Hum. Mol. Genet. 2002, 11, 477–486. [Google Scholar] [CrossRef]
- Hofmann, S.; Rothbauer, U.; Mühlenbein, N.; Neupert, W.; Gerbitz, K.-D.; Brunner, M.; Bauer, M.F. The C66W Mutation in the Deafness Dystonia Peptide 1 (DDP1) Affects the Formation of Functional DDP1.TIM13 Complexes in the Mitochondrial Intermembrane Space. J. Biol. Chem. 2002, 277, 23287–23293. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 Encodes an Inner Mitochondrial Membrane Protein and Is Mutated in Infantile Hepatic Mitochondrial DNA Depletion. Nat. Genet. 2006, 38, 570–575. [Google Scholar] [CrossRef]
- Gilberti, M.; Baruffini, E.; Donnini, C.; Dallabona, C. Pathological Alleles of MPV17 Modeled in the Yeast Saccharomyces cerevisiae Orthologous Gene SYM1 Reveal Their Inability to Take Part in a High Molecular Weight Complex. PLoS ONE 2018, 13, e0205014. [Google Scholar] [CrossRef]
- Stuart, G.R.; Santos, J.H.; Strand, M.K.; Van Houten, B.; Copeland, W.C. Mitochondrial and Nuclear DNA Defects in Saccharomyces cerevisiae with Mutations in DNA Polymerase Gamma Associated with Progressive External Ophthalmoplegia. Hum. Mol. Genet. 2006, 15, 363–374. [Google Scholar] [CrossRef]
- Baruffini, E.; Lodi, T.; Dallabona, C.; Puglisi, A.; Zeviani, M.; Ferrero, I. Genetic and Chemical Rescue of the Saccharomyces cerevisiae Phenotype Induced by Mitochondrial DNA Polymerase Mutations Associated with Progressive External Ophthalmoplegia in Humans. Hum. Mol. Genet. 2006, 15, 2846–2855. [Google Scholar] [CrossRef]
- Baruffini, E.; Ferrero, I.; Foury, F. Mitochondrial DNA Defects in Saccharomyces cerevisiae Caused by Functional Interactions between DNA Polymerase Gamma Mutations Associated with Disease in Human. Biochim. Biophys. Acta 2007, 1772, 1225–1235. [Google Scholar] [CrossRef][Green Version]
- Szczepanowska, K.; Foury, F. A Cluster of Pathogenic Mutations in the 3’-5’ Exonuclease Domain of DNA Polymerase Gamma Defines a Novel Module Coupling DNA Synthesis and Degradation. Hum. Mol. Genet. 2010, 19, 3516–3529. [Google Scholar] [CrossRef]
- Stricker, S.; Prüss, H.; Horvath, R.; Baruffini, E.; Lodi, T.; Siebert, E.; Endres, M.; Zschenderlein, R.; Meisel, A. A Variable Neurodegenerative Phenotype with Polymerase Gamma Mutation. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1181–1182. [Google Scholar] [CrossRef][Green Version]
- Stumpf, J.D.; Bailey, C.M.; Spell, D.; Stillwagon, M.; Anderson, K.S.; Copeland, W.C. Mip1 Containing Mutations Associated with Mitochondrial Disease Causes Mutagenesis and Depletion of MtDNA in Saccharomyces cerevisiae. Hum. Mol. Genet. 2010, 19, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.D.; Horvath, R.; Baruffini, E.; Ferrero, I.; Bulst, S.; Watkins, P.B.; Fontana, R.J.; Day, C.P.; Chinnery, P.F. Polymerase γ Gene POLG Determines the Risk of Sodium Valproate-Induced Liver Toxicity. Hepatology 2010, 52, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Baruffini, E.; Horvath, R.; Dallabona, C.; Czermin, B.; Lamantea, E.; Bindoff, L.; Invernizzi, F.; Ferrero, I.; Zeviani, M.; Lodi, T. Predicting the Contribution of Novel POLG Mutations to Human Disease through Analysis in Yeast Model. Mitochondrion 2011, 11, 182–190. [Google Scholar] [CrossRef]
- Baruffini, E.; Serafini, F.; Ferrero, I.; Lodi, T. Overexpression of DNA Polymerase Zeta Reduces the Mitochondrial Mutability Caused by Pathological Mutations in DNA Polymerase Gamma in Yeast. PLoS ONE 2012, 7, e34322. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, J.D.; Copeland, W.C. The Exonuclease Activity of the Yeast Mitochondrial DNA Polymerase γ Suppresses Mitochondrial DNA Deletions between Short Direct Repeats in Saccharomyces cerevisiae. Genetics 2013, 194, 519–522. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stumpf, J.D.; Copeland, W.C. MMS Exposure Promotes Increased MtDNA Mutagenesis in the Presence of Replication-Defective Disease-Associated DNA Polymerase γ Variants. PLoS Genet. 2014, 10, e1004748. [Google Scholar] [CrossRef]
- Kaliszewska, M.; Kruszewski, J.; Kierdaszuk, B.; Kostera-Pruszczyk, A.; Nojszewska, M.; Łusakowska, A.; Vizueta, J.; Sabat, D.; Lutyk, D.; Lower, M.; et al. Yeast Model Analysis of Novel Polymerase Gamma Variants Found in Patients with Autosomal Recessive Mitochondrial Disease. Hum. Genet. 2015, 134, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Hoyos-Gonzalez, N.; Trasviña-Arenas, C.H.; Degiorgi, A.; Castro-Lara, A.Y.; Peralta-Castro, A.; Jimenez-Sandoval, P.; Diaz-Quezada, C.; Lodi, T.; Baruffini, E.; Brieba, L.G. Modeling of Pathogenic Variants of Mitochondrial DNA Polymerase: Insight into the Replication Defects and Implication for Human Disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129608. [Google Scholar] [CrossRef]
- Qian, Y.; Kachroo, A.H.; Yellman, C.M.; Marcotte, E.M.; Johnson, K.A. Yeast Cells Expressing the Human Mitochondrial DNA Polymerase Reveal Correlations between Polymerase Fidelity and Human Disease Progression. J. Biol. Chem. 2014, 289, 5970–5985. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Ziehr, J.L.; Johnson, K.A. Alpers Disease Mutations in Human DNA Polymerase Gamma Cause Catalytic Defects in Mitochondrial DNA Replication by Distinct Mechanisms. Front. Genet. 2015, 6, 135. [Google Scholar] [CrossRef]
- Haijes, H.A.; Koster, M.J.E.; Rehmann, H.; Li, D.; Hakonarson, H.; Cappuccio, G.; Hancarova, M.; Lehalle, D.; Reardon, W.; Schaefer, G.B.; et al. De Novo Heterozygous POLR2A Variants Cause a Neurodevelopmental Syndrome with Profound Infantile-Onset Hypotonia. Am. J. Hum. Genet. 2019, 105, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Kopajtich, R.; Freisinger, P.; Wieland, T.; Rorbach, J.; Nicholls, T.J.; Baruffini, E.; Walther, A.; Danhauser, K.; Zimmermann, F.A.; et al. ELAC2 Mutations Cause a Mitochondrial RNA Processing Defect Associated with Hypertrophic Cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guan, M.-X. A Human Mitochondrial GTP Binding Protein Related to TRNA Modification May Modulate Phenotypic Expression of the Deafness-Associated Mitochondrial 12S RRNA Mutation. Mol. Cell. Biol. 2002, 22, 7701–7711. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; D’Souza, A.R.; Dallabona, C.; Lodi, T.; Rebelo-Guiomar, P.; Rorbach, J.; Donati, M.A.; Procopio, E.; Montomoli, M.; Guerrini, R.; et al. Defective Mitochondrial RRNA Methyltransferase MRM2 Causes MELAS-like Clinical Syndrome. Hum. Mol. Genet. 2017, 26, 4257–4266. [Google Scholar] [CrossRef]
- Baruffini, E.; Dallabona, C.; Invernizzi, F.; Yarham, J.W.; Melchionda, L.; Blakely, E.L.; Lamantea, E.; Donnini, C.; Santra, S.; Vijayaraghavan, S.; et al. MTO1 Mutations Are Associated with Hypertrophic Cardiomyopathy and Lactic Acidosis and Cause Respiratory Chain Deficiency in Humans and Yeast. Hum. Mutat. 2013, 34, 1501–1509. [Google Scholar] [CrossRef]
- Ghezzi, D.; Baruffini, E.; Haack, T.B.; Invernizzi, F.; Melchionda, L.; Dallabona, C.; Strom, T.M.; Parini, R.; Burlina, A.B.; Meitinger, T.; et al. Mutations of the Mitochondrial-TRNA Modifier MTO1 Cause Hypertrophic Cardiomyopathy and Lactic Acidosis. Am. J. Hum. Genet. 2012, 90, 1079–1087. [Google Scholar] [CrossRef]
- Yarham, J.W.; Lamichhane, T.N.; Pyle, A.; Mattijssen, S.; Baruffini, E.; Bruni, F.; Donnini, C.; Vassilev, A.; He, L.; Blakely, E.L.; et al. Defective I6A37 Modification of Mitochondrial and Cytosolic TRNAs Results from Pathogenic Mutations in TRIT1 and Its Substrate TRNA. PLoS Genet. 2014, 10, e1004424. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.A.; Kopajtich, R.; D’Souza, A.R.; Rorbach, J.; Kremer, L.S.; Husain, R.A.; Dallabona, C.; Donnini, C.; Alston, C.L.; Griffin, H.; et al. TRMT5 Mutations Cause a Defect in Post-Transcriptional Modification of Mitochondrial TRNA Associated with Multiple Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2015, 97, 319–328. [Google Scholar] [CrossRef]
- Yan, Q.; Li, X.; Faye, G.; Guan, M.-X. Mutations in MTO2 Related to TRNA Modification Impair Mitochondrial Gene Expression and Protein Synthesis in the Presence of a Paromomycin Resistance Mutation in Mitochondrial 15 S RRNA. J. Biol. Chem. 2005, 280, 29151–29157. [Google Scholar] [CrossRef] [PubMed]
- Umeda, N.; Suzuki, T.; Yukawa, M.; Ohya, Y.; Shindo, H.; Watanabe, K.; Suzuki, T. Mitochondria-Specific RNA-Modifying Enzymes Responsible for the Biosynthesis of the Wobble Base in Mitochondrial TRNAs. Implications for the Molecular Pathogenesis of Human Mitochondrial Diseases. J. Biol. Chem. 2005, 280, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.K.; Schmitz-Abe, K.; Kennedy, E.K.; Mamady, H.; Naas, T.; Durie, D.; Campagna, D.R.; Lau, A.; Sendamarai, A.K.; Wiseman, D.H.; et al. Mutations in TRNT1 Cause Congenital Sideroblastic Anemia with Immunodeficiency, Fevers, and Developmental Delay (SIFD). Blood 2014, 124, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- Leibovitch, M.; Reid, N.E.; Victoria, J.; Hanic-Joyce, P.J.; Joyce, P.B.M. Analysis of the Pathogenic I326T Variant of Human TRNA Nucleotidyltransferase Reveals Reduced Catalytic Activity and Thermal Stability in Vitro Linked to a Conformational Change. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 616–626. [Google Scholar] [CrossRef]
- Dallabona, C.; Diodato, D.; Kevelam, S.H.; Haack, T.B.; Wong, L.-J.; Salomons, G.S.; Baruffini, E.; Melchionda, L.; Mariotti, C.; Strom, T.M.; et al. Novel (Ovario) Leukodystrophy Related to AARS2 Mutations. Neurology 2014, 82, 2063–2071. [Google Scholar] [CrossRef]
- Kuo, M.E.; Antonellis, A.; Shakkottai, V.G. Alanyl-TRNA Synthetase 2 (AARS2)-Related Ataxia Without Leukoencephalopathy. Cerebellum 2020, 19, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Griffin, L.B.; Sakaguchi, R.; McGuigan, D.; Gonzalez, M.A.; Searby, C.; Züchner, S.; Hou, Y.-M.; Antonellis, A. Impaired Function Is a Common Feature of Neuropathy-Associated Glycyl-TRNA Synthetase Mutations. Hum. Mutat 2014, 35, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Meyer-Schuman, R.; Bacon, C.; Shy, M.E.; Antonellis, A.; Scherer, S.S. A Recurrent GARS Mutation Causes Distal Hereditary Motor Neuropathy. J. Peripher. Nerv. Syst. 2019, 24, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Markovitz, R.; Ghosh, R.; Kuo, M.E.; Hong, W.; Lim, J.; Bernes, S.; Manberg, S.; Crosby, K.; Tanpaiboon, P.; Bharucha-Goebel, D.; et al. GARS-Related Disease in Infantile Spinal Muscular Atrophy: Implications for Diagnosis and Treatment. Am. J. Med. Genet. A 2020, 182, 1167–1176. [Google Scholar] [CrossRef]
- Friederich, M.W.; Timal, S.; Powell, C.A.; Dallabona, C.; Kurolap, A.; Palacios-Zambrano, S.; Bratkovic, D.; Derks, T.G.J.; Bick, D.; Bouman, K.; et al. Pathogenic Variants in Glutamyl-TRNAGln Amidotransferase Subunits Cause a Lethal Mitochondrial Cardiomyopathy Disorder. Nat. Commun. 2018, 9, 4065. [Google Scholar] [CrossRef]
- Pierce, S.B.; Chisholm, K.M.; Lynch, E.D.; Lee, M.K.; Walsh, T.; Opitz, J.M.; Li, W.; Klevit, R.E.; King, M.-C. Mutations in Mitochondrial Histidyl TRNA Synthetase HARS2 Cause Ovarian Dysgenesis and Sensorineural Hearing Loss of Perrault Syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 6543–6548. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, J.-B.; Zeng, Q.-Y.; Wu, S.; Xue, M.-Q.; Fang, P.; Wang, E.-D.; Zhou, X.-L. Hearing Impairment-Associated KARS Mutations Lead to Defects in Aminoacylation of Both Cytoplasmic and Mitochondrial TRNALys. Sci. China Life Sci. 2020, 63, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Gersak, K.; Michaelson-Cohen, R.; Walsh, T.; Lee, M.K.; Malach, D.; Klevit, R.E.; King, M.-C.; Levy-Lahad, E. Mutations in LARS2, Encoding Mitochondrial Leucyl-TRNA Synthetase, Lead to Premature Ovarian Failure and Hearing Loss in Perrault Syndrome. Am. J. Hum. Genet. 2013, 92, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Cassandrini, D.; Cilio, M.R.; Bianchi, M.; Doimo, M.; Balestri, M.; Tessa, A.; Rizza, T.; Sartori, G.; Meschini, M.C.; Nesti, C.; et al. Pontocerebellar Hypoplasia Type 6 Caused by Mutations in RARS2: Definition of the Clinical Spectrum and Molecular Findings in Five Patients. J. Inherit. Metab. Dis. 2013, 36, 43–53. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, X.-L.; Ruan, Z.-R.; Liu, R.-J.; Eriani, G.; Wang, E.-D. A Human Disease-Causing Point Mutation in Mitochondrial Threonyl-TRNA Synthetase Induces Both Structural and Functional Defects. J. Biol. Chem. 2016, 291, 6507–6520. [Google Scholar] [CrossRef]
- Diodato, D.; Melchionda, L.; Haack, T.B.; Dallabona, C.; Baruffini, E.; Donnini, C.; Granata, T.; Ragona, F.; Balestri, P.; Margollicci, M.; et al. VARS2 and TARS2 Mutations in Patients with Mitochondrial Encephalomyopathies. Hum. Mutat. 2014, 35, 983–989. [Google Scholar] [CrossRef]
- Chin, H.-L.; Goh, D.L.-M.; Wang, F.S.; Tay, S.K.H.; Heng, C.K.; Donnini, C.; Baruffini, E.; Pines, O. A Combination of Two Novel VARS2 Variants Causes a Mitochondrial Disorder Associated with Failure to Thrive and Pulmonary Hypertension. J. Mol. Med. 2019, 97, 1557–1566. [Google Scholar] [CrossRef]
- Maffezzini, C.; Laine, I.; Dallabona, C.; Clemente, P.; Calvo-Garrido, J.; Wibom, R.; Naess, K.; Barbaro, M.; Falk, A.; Donnini, C.; et al. Mutations in the Mitochondrial Tryptophanyl-TRNA Synthetase Cause Growth Retardation and Progressive Leukoencephalopathy. Mol. Genet. Genom. Med. 2019, 7, e654. [Google Scholar] [CrossRef] [PubMed]
- Ardissone, A.; Lamantea, E.; Quartararo, J.; Dallabona, C.; Carrara, F.; Moroni, I.; Donnini, C.; Garavaglia, B.; Zeviani, M.; Uziel, G. A Novel Homozygous YARS2 Mutation in Two Italian Siblings and a Review of Literature. JIMD Rep. 2015, 20, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Sommerville, E.W.; Ng, Y.S.; Alston, C.L.; Dallabona, C.; Gilberti, M.; He, L.; Knowles, C.; Chin, S.L.; Schaefer, A.M.; Falkous, G.; et al. Clinical Features, Molecular Heterogeneity, and Prognostic Implications in YARS2-Related Mitochondrial Myopathy. JAMA Neurol. 2017, 74, 686–694. [Google Scholar] [CrossRef]
- Smith, F.; Hopton, S.; Dallabona, C.; Gilberti, M.; Falkous, G.; Norwood, F.; Donnini, C.; Gorman, G.S.; Clark, B.; Taylor, R.W.; et al. Sideroblastic Anemia with Myopathy Secondary to Novel, Pathogenic Missense Variants in the YARS2 Gene. Haematologica 2018, 103, e564–e566. [Google Scholar] [CrossRef]
- Valente, L.; Tiranti, V.; Marsano, R.M.; Malfatti, E.; Fernandez-Vizarra, E.; Donnini, C.; Mereghetti, P.; De Gioia, L.; Burlina, A.; Castellan, C.; et al. Infantile Encephalopathy and Defective Mitochondrial DNA Translation in Patients with Mutations of Mitochondrial Elongation Factors EFG1 and EFTu. Am. J. Hum. Genet. 2007, 80, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Montanari, A.; Zhou, Y.F.; D’Orsi, M.F.; Bolotin-Fukuhara, M.; Frontali, L.; Francisci, S. Analyzing the Suppression of Respiratory Defects in the Yeast Model of Human Mitochondrial TRNA Diseases. Gene 2013, 527, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Di Nottia, M.; Montanari, A.; Verrigni, D.; Oliva, R.; Torraco, A.; Fernandez-Vizarra, E.; Diodato, D.; Rizza, T.; Bianchi, M.; Catteruccia, M.; et al. Novel Mutation in Mitochondrial Elongation Factor EF-Tu Associated to Dysplastic Leukoencephalopathy and Defective Mitochondrial DNA Translation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 961–967. [Google Scholar] [CrossRef]
- Nasca, A.; Legati, A.; Baruffini, E.; Nolli, C.; Moroni, I.; Ardissone, A.; Goffrini, P.; Ghezzi, D. Biallelic Mutations in DNM1L Are Associated with a Slowly Progressive Infantile Encephalopathy. Hum. Mutat. 2016, 37, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Verrigni, D.; Di Nottia, M.; Ardissone, A.; Baruffini, E.; Nasca, A.; Legati, A.; Bellacchio, E.; Fagiolari, G.; Martinelli, D.; Fusco, L.; et al. Clinical-Genetic Features and Peculiar Muscle Histopathology in Infantile DNM1L-Related Mitochondrial Epileptic Encephalopathy. Hum. Mutat. 2019, 40, 601–618. [Google Scholar] [CrossRef]
- Ashrafian, H.; Docherty, L.; Leo, V.; Towlson, C.; Neilan, M.; Steeples, V.; Lygate, C.A.; Hough, T.; Townsend, S.; Williams, D.; et al. A Mutation in the Mitochondrial Fission Gene Dnm1l Leads to Cardiomyopathy. PLoS Genet. 2010, 6, e1001000. [Google Scholar] [CrossRef]
- Amiott, E.A.; Cohen, M.M.; Saint-Georges, Y.; Weissman, A.M.; Shaw, J.M. A Mutation Associated with CMT2A Neuropathy Causes Defects in Fzo1 GTP Hydrolysis, Ubiquitylation, and Protein Turnover. Mol. Biol. Cell 2009, 20, 5026–5035. [Google Scholar] [CrossRef] [PubMed]
- Del Dotto, V.; Fogazza, M.; Musiani, F.; Maresca, A.; Aleo, S.J.; Caporali, L.; La Morgia, C.; Nolli, C.; Lodi, T.; Goffrini, P.; et al. Deciphering OPA1 Mutations Pathogenicity by Combined Analysis of Human, Mouse and Yeast Cell Models. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3496–3514. [Google Scholar] [CrossRef]
- Nolli, C.; Goffrini, P.; Lazzaretti, M.; Zanna, C.; Vitale, R.; Lodi, T.; Baruffini, E. Validation of a MGM1/OPA1 Chimeric Gene for Functional Analysis in Yeast of Mutations Associated with Dominant Optic Atrophy. Mitochondrion 2015, 25, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Valianpour, F.; Chen, S.; Vaz, F.M.; Hakkaart, G.A.; Wanders, R.J.A.; Greenberg, M.L. Aberrant Cardiolipin Metabolism in the Yeast Taz1 Mutant: A Model for Barth Syndrome. Mol. Microbiol. 2004, 51, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Claypool, S.M.; Whited, K.; Srijumnong, S.; Han, X.; Koehler, C.M. Barth Syndrome Mutations That Cause Tafazzin Complex Lability. J. Cell Biol. 2011, 192, 447–462. [Google Scholar] [CrossRef]
- Whited, K.; Baile, M.G.; Currier, P.; Claypool, S.M. Seven Functional Classes of Barth Syndrome Mutation. Hum. Mol. Genet. 2013, 22, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Thorsness, M.K.; Policastro, R.; McGoldrick, L.L.; Hollingsworth, N.M.; Thorsness, P.E.; Neiman, A.M. Yeast Vps13 Promotes Mitochondrial Function and Is Localized at Membrane Contact Sites. Mol. Biol. Cell 2016, 27, 2435–2449. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Flis, K.; Kaminska, J.; Grynberg, M.; Urbanek, A.; Ayscough, K.R.; Zoladek, T. Amino Acid Substitution Equivalent to Human Chorea-Acanthocytosis I2771R in Yeast Vps13 Protein Affects Its Binding to Phosphatidylinositol 3-Phosphate. Hum. Mol. Genet. 2017, 26, 1497–1510. [Google Scholar] [CrossRef]
- Soczewka, P.; Kolakowski, D.; Smaczynska-de Rooij, I.; Rzepnikowska, W.; Ayscough, K.R.; Kaminska, J.; Zoladek, T. Yeast-Model-Based Study Identified Myosin- and Calcium-Dependent Calmodulin Signalling as a Potential Target for Drug Intervention in Chorea-Acanthocytosis. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Benincá, C.; Zanette, V.; Brischigliaro, M.; Johnson, M.; Reyes, A.; do Valle, D.A.; Robinson, A.J.; Degiorgi, A.; Yeates, A.; Telles, B.A.; et al. Mutation in the MICOS Subunit Gene APOO (MIC26) Associated with an X-Linked Recessive Mitochondrial Myopathy, Lactic Acidosis, Cognitive Impairment and Autistic Features. J. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Kaminska, J.; Kabzińska, D.; Kochański, A. Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model. Genes 2020, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Pines, O.; Ta-Shma, A.; Burak, E.; Shaag, A.; Halvardson, J.; Edvardson, S.; Mahajna, M.; Zenvirt, S.; Saada, A.; et al. Infantile Cerebellar-Retinal Degeneration Associated with a Mutation in Mitochondrial Aconitase, ACO2. Am. J. Hum. Genet. 2012, 90, 518–523. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Gerber, S.; Hubert, L.; Delahodde, A.; Chretien, D.; Gérard, X.; Amati-Bonneau, P.; Giacomotto, M.-C.; Boddaert, N.; Kaminska, A.; et al. Mutations in the Tricarboxylic Acid Cycle Enzyme, Aconitase 2, Cause Either Isolated or Syndromic Optic Neuropathy with Encephalopathy and Cerebellar Atrophy. J. Med. Genet. 2014, 51, 834–838. [Google Scholar] [CrossRef]
- Neumann, M.A.-C.; Grossmann, D.; Schimpf-Linzenbold, S.; Dayan, D.; Stingl, K.; Ben-Menachem, R.; Pines, O.; Massart, F.; Delcambre, S.; Ghelfi, J.; et al. Haploinsufficiency Due to a Novel ACO2 Deletion Causes Mitochondrial Dysfunction in Fibroblasts from a Patient with Dominant Optic Nerve Atrophy. Sci. Rep. 2020, 10, 16736. [Google Scholar] [CrossRef]
- Sharkia, R.; Wierenga, K.J.; Kessel, A.; Azem, A.; Bertini, E.; Carrozzo, R.; Torraco, A.; Goffrini, P.; Ceccatelli Berti, C.; McCormick, M.E.; et al. Clinical, Radiological, and Genetic Characteristics of 16 Patients with ACO2 Gene Defects: Delineation of an Emerging Neurometabolic Syndrome. J. Inherit. Metab. Dis. 2019, 42, 264–275. [Google Scholar] [CrossRef]
- Fattal-Valevski, A.; Eliyahu, H.; Fraenkel, N.D.; Elmaliach, G.; Hausman-Kedem, M.; Shaag, A.; Mandel, D.; Pines, O.; Elpeleg, O. Homozygous Mutation, p.Pro304His, in IDH3A, Encoding Isocitrate Dehydrogenase Subunit Is Associated with Severe Encephalopathy in Infancy. Neurogenetics 2017, 18, 57–61. [Google Scholar] [CrossRef]
- Ait-El-Mkadem, S.; Dayem-Quere, M.; Gusic, M.; Chaussenot, A.; Bannwarth, S.; François, B.; Genin, E.C.; Fragaki, K.; Volker-Touw, C.L.M.; Vasnier, C.; et al. Mutations in MDH2, Encoding a Krebs Cycle Enzyme, Cause Early-Onset Severe Encephalopathy. Am. J. Hum. Genet. 2017, 100, 151–159. [Google Scholar] [CrossRef]
- Heimer, G.; Kerätär, J.M.; Riley, L.G.; Balasubramaniam, S.; Eyal, E.; Pietikäinen, L.P.; Hiltunen, J.K.; Marek-Yagel, D.; Hamada, J.; Gregory, A.; et al. MECR Mutations Cause Childhood-Onset Dystonia and Optic Atrophy, a Mitochondrial Fatty Acid Synthesis Disorder. Am. J. Hum. Genet. 2016, 99, 1229–1244. [Google Scholar] [CrossRef] [PubMed]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.-C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila, and Humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Drakulic, S.; Rai, J.; Petersen, S.V.; Golas, M.M.; Sander, B. Folding and Assembly Defects of Pyruvate Dehydrogenase Deficiency-Related Variants in the E1α Subunit of the Pyruvate Dehydrogenase Complex. Cell. Mol. Life Sci. 2018, 75, 3009–3026. [Google Scholar] [CrossRef] [PubMed]
- Aral, B.; Benelli, C.; Ait-Ghezala, G.; Amessou, M.; Fouque, F.; Maunoury, C.; Créau, N.; Kamoun, P.; Marsac, C. Mutations in PDX1, the Human Lipoyl-Containing Component X of the Pyruvate Dehydrogenase-Complex Gene on Chromosome 11p1, in Congenital Lactic Acidosis. Am. J. Hum. Genet. 1997, 61, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Guimier, A.; Gordon, C.T.; Godard, F.; Ravenscroft, G.; Oufadem, M.; Vasnier, C.; Rambaud, C.; Nitschke, P.; Bole-Feysot, C.; Masson, C.; et al. Biallelic PPA2 Mutations Cause Sudden Unexpected Cardiac Arrest in Infancy. Am. J. Hum. Genet. 2016, 99, 666–673. [Google Scholar] [CrossRef]
- Zeng, H.-S.; Zhao, S.-T.; Deng, M.; Zhang, Z.-H.; Cai, X.-R.; Chen, F.-P.; Song, Y.-Z. Inspissated Bile Syndrome in an Infant with Citrin Deficiency and Congenital Anomalies of the Biliary Tract and Esophagus: Identification and Pathogenicity Analysis of a Novel SLC25A13 Mutation with Incomplete Penetrance. Int. J. Mol. Med. 2014, 34, 1241–1248. [Google Scholar] [CrossRef][Green Version]
- Seifert, E.L.; Gál, A.; Acoba, M.G.; Li, Q.; Anderson-Pullinger, L.; Golenár, T.; Moffat, C.; Sondheimer, N.; Claypool, S.M.; Hajnóczky, G. Natural and Induced Mitochondrial Phosphate Carrier Loss: Differential dependence of mitochondrial metabolism and dynamics and cell survival on the extent of depletion. J. Biol. Chem. 2016, 291, 26126–26137. [Google Scholar] [CrossRef]
- Mayr, J.A.; Merkel, O.; Kohlwein, S.D.; Gebhardt, B.R.; Böhles, H.; Fötschl, U.; Koch, J.; Jaksch, M.; Lochmüller, H.; Horváth, R.; et al. Mitochondrial Phosphate-Carrier Deficiency: A Novel Disorder of Oxidative Phosphorylation. Am. J. Hum. Genet. 2007, 80, 478–484. [Google Scholar] [CrossRef]
- Allikmets, R.; Raskind, W.H.; Hutchinson, A.; Schueck, N.D.; Dean, M.; Koeller, D.M. Mutation of a Putative Mitochondrial Iron Transporter Gene (ABC7) in X-Linked Sideroblastic Anemia and Ataxia (XLSA/A). Hum. Mol. Genet. 1999, 8, 743–749. [Google Scholar] [CrossRef]
- Pearson, S.A.; Wachnowsky, C.; Cowan, J.A. Defining the Mechanism of the Mitochondrial Atm1p [2Fe-2S] Cluster Exporter. Metallomics 2020, 12, 902–915. [Google Scholar] [CrossRef]
- Bekri, S.; Kispal, G.; Lange, H.; Fitzsimons, E.; Tolmie, J.; Lill, R.; Bishop, D.F. Human ABC7 Transporter: Gene Structure and Mutation Causing X-Linked Sideroblastic Anemia with Ataxia with Disruption of Cytosolic Iron-Sulfur Protein Maturation. Blood 2000, 96, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Drecourt, A.; Petit, F.; Deguine, D.D.; Vasnier, C.; Oufadem, M.; Masson, C.; Bonnet, C.; Masmoudi, S.; Mosnier, I.; et al. FDXR Mutations Cause Sensorial Neuropathies and Expand the Spectrum of Mitochondrial Fe-S-Synthesis Diseases. Am. J. Hum. Genet. 2017, 101, 630–637. [Google Scholar] [CrossRef]
- Cavadini, P.; Gellera, C.; Patel, P.I.; Isaya, G. Human Frataxin Maintains Mitochondrial Iron Homeostasis in Saccharomyces cerevisiae. Hum. Mol. Genet. 2000, 9, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.B.; Roof, D.M. Respiratory Deficiency Due to Loss of Mitochondrial DNA in Yeast Lacking the Frataxin Homologue. Nat. Genet. 1997, 16, 352–357. [Google Scholar] [CrossRef]
- Foury, F. Screens for Mitochondrial Mutants in Yeast. Methods Mol. Biol. 2007, 372, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-M.; Narayanan, V.; Mieczkowski, P.A.; Petes, T.D.; Krasilnikova, M.M.; Mirkin, S.M.; Lobachev, K.S. Chromosome Fragility at GAA Tracts in Yeast Depends on Repeat Orientation and Requires Mismatch Repair. EMBO J. 2008, 27, 2896–2906. [Google Scholar] [CrossRef]
- Leidgens, S.; De Smet, S.; Foury, F. Frataxin Interacts with Isu1 through a Conserved Tryptophan in Its Beta-Sheet. Hum. Mol. Genet. 2010, 19, 276–286. [Google Scholar] [CrossRef][Green Version]
- Khristich, A.N.; Armenia, J.F.; Matera, R.M.; Kolchinski, A.A.; Mirkin, S.M. Large-Scale Contractions of Friedreich’s Ataxia GAA Repeats in Yeast Occur during DNA Replication Due to Their Triplex-Forming Ability. Proc. Natl. Acad. Sci. USA 2020, 117, 1628–1637. [Google Scholar] [CrossRef]
- Saha, P.P.; Kumar, S.K.P.; Srivastava, S.; Sinha, D.; Pareek, G.; D’Silva, P. The Presence of Multiple Cellular Defects Associated with a Novel G50E Iron-Sulfur Cluster Scaffold Protein (ISCU) Mutation Leads to Development of Mitochondrial Myopathy. J. Biol. Chem. 2014, 289, 10359–10377. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Reyes, A.; Ceccatelli Berti, C.; Stehling, O.; Marchet, S.; Lamperti, C.; Ferrari, A.; Robinson, A.J.; Mühlenhoff, U.; Lill, R.; et al. A Novel de Novo Dominant Mutation in ISCU Associated with Mitochondrial Myopathy. J. Med. Genet. 2017, 54, 815–824. [Google Scholar] [CrossRef]
- Lim, S.C.; Friemel, M.; Marum, J.E.; Tucker, E.J.; Bruno, D.L.; Riley, L.G.; Christodoulou, J.; Kirk, E.P.; Boneh, A.; DeGennaro, C.M.; et al. Mutations in LYRM4, Encoding Iron-Sulfur Cluster Biogenesis Factor ISD11, Cause Deficiency of Multiple Respiratory Chain Complexes. Hum. Mol. Genet. 2013, 22, 4460–4473. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; Del Toro, M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J.; et al. A Fatal Mitochondrial Disease Is Associated with Defective NFU1 Function in the Maturation of a Subset of Mitochondrial Fe-S Proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Dusi, S.; Valletta, L.; Haack, T.B.; Tsuchiya, Y.; Venco, P.; Pasqualato, S.; Goffrini, P.; Tigano, M.; Demchenko, N.; Wieland, T.; et al. Exome Sequence Reveals Mutations in CoA Synthase as a Cause of Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2014, 94, 11–22. [Google Scholar] [CrossRef]
- Ceccatelli Berti, C.; Dallabona, C.; Lazzaretti, M.; Dusi, S.; Tosi, E.; Tiranti, V.; Goffrini, P. Modeling Human Coenzyme A Synthase Mutation in Yeast Reveals Altered Mitochondrial Function, Lipid Content and Iron Metabolism. Microb. Cell 2015, 2, 126–135. [Google Scholar] [CrossRef]
- Soreze, Y.; Boutron, A.; Habarou, F.; Barnerias, C.; Nonnenmacher, L.; Delpech, H.; Mamoune, A.; Chrétien, D.; Hubert, L.; Bole-Feysot, C.; et al. Mutations in Human Lipoyltransferase Gene LIPT1 Cause a Leigh Disease with Secondary Deficiency for Pyruvate and Alpha-Ketoglutarate Dehydrogenase. Orphanet J. Rare Dis. 2013, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Habarou, F.; Hamel, Y.; Haack, T.B.; Feichtinger, R.G.; Lebigot, E.; Marquardt, I.; Busiah, K.; Laroche, C.; Madrange, M.; Grisel, C.; et al. Biallelic Mutations in LIPT2 Cause a Mitochondrial Lipoylation Defect Associated with Severe Neonatal Encephalopathy. Am. J. Hum. Genet. 2017, 101, 283–290. [Google Scholar] [CrossRef]
- Ceccatelli Berti, C.; Gilea, A.I.; De Gregorio, M.A.; Goffrini, P. Exploring Yeast as a Study Model of Pantothenate Kinase-Associated Neurodegeneration and for the Identification of Therapeutic Compounds. Int. J. Mol. Sci 2020, 22, 293. [Google Scholar] [CrossRef]
- Edvardson, S.; Porcelli, V.; Jalas, C.; Soiferman, D.; Kellner, Y.; Shaag, A.; Korman, S.H.; Pierri, C.L.; Scarcia, P.; Fraenkel, N.D.; et al. Agenesis of Corpus Callosum and Optic Nerve Hypoplasia Due to Mutations in SLC25A1 Encoding the Mitochondrial Citrate Transporter. J. Med. Genet. 2013, 50, 240–245. [Google Scholar] [CrossRef]
- Palmieri, L.; Alberio, S.; Pisano, I.; Lodi, T.; Meznaric-Petrusa, M.; Zidar, J.; Santoro, A.; Scarcia, P.; Fontanesi, F.; Lamantea, E.; et al. Complete Loss-of-Function of the Heart/Muscle-Specific Adenine Nucleotide Translocator Is Associated with Mitochondrial Myopathy and Cardiomyopathy. Hum. Mol. Genet. 2005, 14, 3079–3088. [Google Scholar] [CrossRef]
- Chen, X.J. Induction of an Unregulated Channel by Mutations in Adenine Nucleotide Translocase Suggests an Explanation for Human Ophthalmoplegia. Hum. Mol. Genet. 2002, 11, 1835–1843. [Google Scholar] [CrossRef]
- Fontanesi, F.; Palmieri, L.; Scarcia, P.; Lodi, T.; Donnini, C.; Limongelli, A.; Tiranti, V.; Zeviani, M.; Ferrero, I.; Viola, A.M. Mutations in AAC2, Equivalent to Human AdPEO-Associated ANT1 Mutations, Lead to Defective Oxidative Phosphorylation in Saccharomyces cerevisiae and Affect Mitochondrial DNA Stability. Hum. Mol. Genet. 2004, 13, 923–934. [Google Scholar] [CrossRef]
- Lodi, T.; Bove, C.; Fontanesi, F.; Viola, A.M.; Ferrero, I. Mutation D104G in ANT1 Gene: Complementation Study in Saccharomyces cerevisiae as a Model System. Biochem. Biophys Res. Commun. 2006, 341, 810–815. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Chen, X.J. Misfolding of Mutant Adenine Nucleotide Translocase in Yeast Supports a Novel Mechanism of Ant1-Induced Muscle Diseases. Mol. Biol. Cell 2015, 26, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Salinas, K.; Zuo, X.; Kucejova, B.; Chen, X.J. Dominant Membrane Uncoupling by Mutant Adenine Nucleotide Translocase in Mitochondrial Diseases. Hum. Mol. Genet. 2008, 17, 4036–4044. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.P.; Keränen, S.; Peltonen, L.; Suomalainen, A. Role of Adenine Nucleotide Translocator 1 in MtDNA Maintenance. Science 2000, 289, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Dallabona, C.; Baruffini, E.; Goffrini, P.; Lodi, T. Dominance of Yeast Aac2R96H and Aac2R252G Mutations, Equivalent to Pathological Mutations in Ant1, Is Due to Gain of Function. Biochem. Biophys Res. Commun. 2017, 493, 909–913. [Google Scholar] [CrossRef]
- Thompson, K.; Majd, H.; Dallabona, C.; Reinson, K.; King, M.S.; Alston, C.L.; He, L.; Lodi, T.; Jones, S.A.; Fattal-Valevski, A.; et al. Recurrent De Novo Dominant Mutations in SLC25A4 Cause Severe Early-Onset Mitochondrial Disease and Loss of Mitochondrial DNA Copy Number. Am. J. Hum. Genet. 2016, 99, 860–876. [Google Scholar] [CrossRef]
- Spiegel, R.; Shaag, A.; Edvardson, S.; Mandel, H.; Stepensky, P.; Shalev, S.A.; Horovitz, Y.; Pines, O.; Elpeleg, O. SLC25A19 Mutation as a Cause of Neuropathy and Bilateral Striatal Necrosis. Ann. Neurol. 2009, 66, 419–424. [Google Scholar] [CrossRef]
- Schiff, M.; Veauville-Merllié, A.; Su, C.H.; Tzagoloff, A.; Rak, M.; Ogier de Baulny, H.; Boutron, A.; Smedts-Walters, H.; Romero, N.B.; Rigal, O.; et al. SLC25A32 Mutations and Riboflavin-Responsive Exercise Intolerance. N. Engl. J. Med. 2016, 374, 795–797. [Google Scholar] [CrossRef]
- Ghosh, A.; Trivedi, P.P.; Timbalia, S.A.; Griffin, A.T.; Rahn, J.J.; Chan, S.S.L.; Gohil, V.M. Copper Supplementation Restores Cytochrome c Oxidase Assembly Defect in a Mitochondrial Disease Model of COA6 Deficiency. Hum. Mol. Genet. 2014, 23, 3596–3606. [Google Scholar] [CrossRef]
- Ghosh, A.; Pratt, A.T.; Soma, S.; Theriault, S.G.; Griffin, A.T.; Trivedi, P.P.; Gohil, V.M. Mitochondrial Disease Genes COA6, COX6B and SCO2 Have Overlapping Roles in COX2 Biogenesis. Hum. Mol. Genet. 2016, 25, 660–671. [Google Scholar] [CrossRef] [PubMed]
- López-Martín, J.M.; Salviati, L.; Trevisson, E.; Montini, G.; DiMauro, S.; Quinzii, C.; Hirano, M.; Rodriguez-Hernandez, A.; Cordero, M.D.; Sánchez-Alcázar, J.A.; et al. Missense Mutation of the COQ2 Gene Causes Defects of Bioenergetics and de Novo Pyrimidine Synthesis. Hum. Mol. Genet. 2007, 16, 1091–1097. [Google Scholar] [CrossRef]
- Multiple-System Atrophy Research Collaboration Mutations in COQ2 in Familial and Sporadic Multiple-System Atrophy. N. Engl. J. Med. 2013, 369, 233–244. [CrossRef]
- Desbats, M.A.; Morbidoni, V.; Silic-Benussi, M.; Doimo, M.; Ciminale, V.; Cassina, M.; Sacconi, S.; Hirano, M.; Basso, G.; Pierrel, F.; et al. The COQ2 Genotype Predicts the Severity of Coenzyme Q10 Deficiency. Hum. Mol. Genet. 2016, 25, 4256–4265. [Google Scholar] [CrossRef]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; de Lonlay, P.; Munnich, A.; Rötig, A. Prenyldiphosphate Synthase, Subunit 1 (PDSS1) and OH-Benzoate Polyprenyltransferase (COQ2) Mutations in Ubiquinone Deficiency and Oxidative Phosphorylation Disorders. J. Clin. Invest. 2007, 117, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Rodriguez Hernandez, M.A.; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency of COQ4 Causes Coenzyme Q10 Deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Bürgi, S.; et al. COQ4 Mutations Cause a Broad Spectrum of Mitochondrial Disorders Associated with CoQ10 Deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.P.T.; Casarin, A.; Desbats, M.A.; Doimo, M.; Trevisson, E.; Santos-Ocaña, C.; Navas, P.; Clarke, C.F.; Salviati, L. Molecular Characterization of the Human COQ5 C-Methyltransferase in Coenzyme Q10 Biosynthesis. Biochim. Biophys. Acta 2014, 1841, 1628–1638. [Google Scholar] [CrossRef]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 Mutations in Human Patients Produce Nephrotic Syndrome with Sensorineural Deafness. J. Clin. Invest. 2011, 121, 2013–2024. [Google Scholar] [CrossRef]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of Vanillic Acid on COQ6 Mutants Identified in Patients with Coenzyme Q10 Deficiency. Biochim. Biophys. Acta 2014, 1842, 1–6. [Google Scholar] [CrossRef]
- Xie, L.X.; Hsieh, E.J.; Watanabe, S.; Allan, C.M.; Chen, J.Y.; Tran, U.C.; Clarke, C.F. Expression of the Human Atypical Kinase ADCK3 Rescues Coenzyme Q Biosynthesis and Phosphorylation of Coq Polypeptides in Yeast Coq8 Mutants. Biochim. Biophys. Acta 2011, 1811, 348–360. [Google Scholar] [CrossRef]
- Vazquez Fonseca, L.; Doimo, M.; Calderan, C.; Desbats, M.A.; Acosta, M.J.; Cerqua, C.; Cassina, M.; Ashraf, S.; Hildebrandt, F.; Sartori, G.; et al. Mutations in COQ8B (ADCK4) Found in Patients with Steroid-Resistant Nephrotic Syndrome Alter COQ8B Function. Hum. Mutat 2018, 39, 406–414. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Black, D.S.; Allan, C.M.; Meunier, B.; Rahman, S.; Clarke, C.F. Human COQ9 Rescues a Coq9 Yeast Mutant by Enhancing Coenzyme Q Biosynthesis from 4-Hydroxybenzoic Acid and Stabilizing the CoQ-Synthome. Front. Physiol. 2017, 8, 463. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; López, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A Nonsense Mutation in COQ9 Causes Autosomal-Recessive Neonatal-Onset Primary Coenzyme Q10 Deficiency: A Potentially Treatable Form of Mitochondrial Disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef]
- De Rocco, D.; Cerqua, C.; Goffrini, P.; Russo, G.; Pastore, A.; Meloni, F.; Nicchia, E.; Moraes, C.T.; Pecci, A.; Salviati, L.; et al. Mutations of Cytochrome c Identified in Patients with Thrombocytopenia THC4 Affect Both Apoptosis and Cellular Bioenergetics. Biochim. Biophys. Acta 2014, 1842, 269–274. [Google Scholar] [CrossRef]
- Uchiyama, Y.; Yanagisawa, K.; Kunishima, S.; Shiina, M.; Ogawa, Y.; Nakashima, M.; Hirato, J.; Imagawa, E.; Fujita, A.; Hamanaka, K.; et al. A Novel CYCS Mutation in the α-Helix of the CYCS C-Terminal Domain Causes Non-Syndromic Thrombocytopenia. Clin. Genet. 2018, 94, 548–553. [Google Scholar] [CrossRef]
- Wimplinger, I.; Morleo, M.; Rosenberger, G.; Iaconis, D.; Orth, U.; Meinecke, P.; Lerer, I.; Ballabio, A.; Gal, A.; Franco, B.; et al. Mutations of the Mitochondrial Holocytochrome C-Type Synthase in X-Linked Dominant Microphthalmia with Linear Skin Defects Syndrome. Am. J. Hum. Genet. 2006, 79, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Wimplinger, I.; Shaw, G.M.; Kutsche, K. HCCS Loss-of-Function Missense Mutation in a Female with Bilateral Microphthalmia and Sclerocornea: A Novel Gene for Severe Ocular Malformations? Mol. Vis. 2007, 13, 1475–1482. [Google Scholar]
- Indrieri, A.; Conte, I.; Chesi, G.; Romano, A.; Quartararo, J.; Tatè, R.; Ghezzi, D.; Zeviani, M.; Goffrini, P.; Ferrero, I.; et al. The Impairment of HCCS Leads to MLS Syndrome by Activating a Non-Canonical Cell Death Pathway in the Brain and Eyes. EMBO Mol. Med. 2013, 5, 280–293. [Google Scholar] [CrossRef]
- Cruciat, C.M.; Hell, K.; Fölsch, H.; Neupert, W.; Stuart, R.A. Bcs1p, an AAA-Family Member, Is a Chaperone for the Assembly of the Cytochrome Bc(1) Complex. EMBO J. 1999, 18, 5226–5233. [Google Scholar] [CrossRef]
- Aghajanian, S.; Worrall, D.M. Identification and Characterization of the Gene Encoding the Human Phosphopantetheine Adenylyltransferase and Dephospho-CoA Kinase Bifunctional Enzyme (CoA Synthase). Biochem. J. 2002, 365, 13–18. [Google Scholar] [CrossRef]
- Di Meo, I.; Cavestro, C.; Pedretti, S.; Fu, T.; Ligorio, S.; Manocchio, A.; Lavermicocca, L.; Santambrogio, P.; Ripamonti, M.; Levi, S.; et al. Neuronal Ablation of CoA Synthase Causes Motor Deficits, Iron Dyshomeostasis, and Mitochondrial Dysfunctions in a CoPAN Mouse Model. Int. J. Mol. Sci. 2020, 21, 9707. [Google Scholar] [CrossRef]
- Storici, F.; Resnick, M.A. Delitto Perfetto Targeted Mutagenesis in Yeast with Oligonucleotides. Genet. Eng. 2003, 25, 189–207. [Google Scholar]
- Olsson, A.; Lind, L.; Thornell, L.-E.; Holmberg, M. Myopathy with Lactic Acidosis Is Linked to Chromosome 12q23.3-24.11 and Caused by an Intron Mutation in the ISCU Gene Resulting in a Splicing Defect. Hum. Mol. Genet. 2008, 17, 1666–1672. [Google Scholar] [CrossRef]
- Mochel, F.; Knight, M.A.; Tong, W.-H.; Hernandez, D.; Ayyad, K.; Taivassalo, T.; Andersen, P.M.; Singleton, A.; Rouault, T.A.; Fischbeck, K.H.; et al. Splice Mutation in the Iron-Sulfur Cluster Scaffold Protein ISCU Causes Myopathy with Exercise Intolerance. Am. J. Hum. Genet. 2008, 82, 652–660. [Google Scholar] [CrossRef]
- Kollberg, G.; Tulinius, M.; Melberg, A.; Darin, N.; Andersen, O.; Holmgren, D.; Oldfors, A.; Holme, E. Clinical Manifestation and a New ISCU Mutation in Iron-Sulphur Cluster Deficiency Myopathy. Brain 2009, 132, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, Y.; Tsuji, J.; Fu, S.-C.; Tomii, K.; Horton, P.; Imai, K. MitoFates: Improved Prediction of Mitochondrial Targeting Sequences and Their Cleavage Sites. Mol. Cell. Proteom. 2015, 14, 1113–1126. [Google Scholar] [CrossRef]
- Nasca, A.; Rizza, T.; Doimo, M.; Legati, A.; Ciolfi, A.; Diodato, D.; Calderan, C.; Carrara, G.; Lamantea, E.; Aiello, C.; et al. Not Only Dominant, Not Only Optic Atrophy: Expanding the Clinical Spectrum Associated with OPA1 Mutations. Orphanet J. Rare Dis. 2017, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, T.; Takemoto, Y.; Hashimoto, M.; Majima, E.; Shinohara, Y.; Terada, H. Significant Expression of Functional Human Type 1 Mitochondrial ADP/ATP Carrier in Yeast Mitochondria. Biol. Pharm. Bull. 2001, 24, 595–599. [Google Scholar] [CrossRef]
- Baruffini, E.; Ferrero, I.; Foury, F. In Vivo Analysis of MtDNA Replication Defects in Yeast. Methods 2010, 51, 426–436. [Google Scholar] [CrossRef]
- Lodi, T.; Dallabona, C.; Nolli, C.; Goffrini, P.; Donnini, C.; Baruffini, E. DNA Polymerase γ and Disease: What We Have Learned from Yeast. Front. Genet. 2015, 6, 106. [Google Scholar] [CrossRef]
- Zakharov, I.A.; Yarovoy, B.P. Cytoduction as a New Tool in Studying the Cytoplasmic Heredity in Yeast. Mol. Cell. Biochem. 1977, 14, 15–18. [Google Scholar] [CrossRef]
- Foury, F.; Szczepanowska, K. Antimutator Alleles of Yeast DNA Polymerase Gamma Modulate the Balance between DNA Synthesis and Excision. PLoS ONE 2011, 6, e27847. [Google Scholar] [CrossRef]
- Barrientos, A.; Fontanesi, F.; Díaz, F. Evaluation of the Mitochondrial Respiratory Chain and Oxidative Phosphorylation System Using Polarography and Spectrophotometric Enzyme Assays. Curr. Protoc Hum. Genet. 2009, 63, 19.3. 1–19.3. 14. [Google Scholar] [CrossRef]
- Somlo, M. Induction and Repression of Mitochondrial ATPase in Yeast. Eur. J. Biochem. 1968, 5, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Lu, K.; Reichert, A.S. Mitophagy and Mitochondrial Dynamics in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2015, 1853, 2766–2774. [Google Scholar] [CrossRef]
- Dujon, B.; Slonimski, P.P.; Weill, L. Mitochondrial Genetics IX: A Model for Recombination and Segregation of Mitochondrial Genomes in Saccharomyces cerevisiae. Genetics 1974, 78, 415–437. [Google Scholar] [CrossRef] [PubMed]
- Hori, A.; Yoshida, M.; Shibata, T.; Ling, F. Reactive Oxygen Species Regulate DNA Copy Number in Isolated Yeast Mitochondria by Triggering Recombination-Mediated Replication. Nucleic Acids Res. 2009, 37, 749–761. [Google Scholar] [CrossRef]
- Chen, X.J.; Butow, R.A. The Organization and Inheritance of the Mitochondrial Genome. Nat. Rev. Genet. 2005, 6, 815–825. [Google Scholar] [CrossRef]
- Lipinski, K.A.; Kaniak-Golik, A.; Golik, P. Maintenance and Expression of the S. Cerevisiae Mitochondrial Genome--from Genetics to Evolution and Systems Biology. Biochim. Biophys. Acta 2010, 1797, 1086–1098. [Google Scholar] [CrossRef]
- Kucej, M.; Butow, R.A. Evolutionary Tinkering with Mitochondrial Nucleoids. Trends Cell Biol. 2007, 17, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Diffley, J.F.; Stillman, B. A Close Relative of the Nuclear, Chromosomal High-Mobility Group Protein HMG1 in Yeast Mitochondria. Proc. Natl. Acad. Sci. USA 1991, 88, 7864–7868. [Google Scholar] [CrossRef]
- Solieri, L. Mitochondrial Inheritance in Budding Yeasts: Towards an Integrated Understanding. Trends Microbiol. 2010, 18, 521–530. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial Inheritance in Yeast. Biochim. Biophys. Acta 2014, 1837, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, I. Organization and Dynamics of Yeast Mitochondrial Nucleoids. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 339–359. [Google Scholar] [CrossRef]
- Sherman, F. Respiration-Deficient Mutants of Yeast. I. Genetics. Genetics 1963, 48, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Dujon, B. Mitochondrial Genetics Revisited. Yeast 2020, 37, 191–205. [Google Scholar] [CrossRef]
- Contamine, V.; Picard, M. Maintenance and Integrity of the Mitochondrial Genome: A Plethora of Nuclear Genes in the Budding Yeast. Microbiol. Mol. Biol. Rev. 2000, 64, 281–315. [Google Scholar] [CrossRef]
- Dujon, B. Mitochondrial genetics and functions. In The Molecular Biology of the Yeast Saccharomyces. Life Cycle and Inheritance; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 1981; pp. 505–635. [Google Scholar]
- Birky, C.W. The Inheritance of Genes in Mitochondria and Chloroplasts: Laws, Mechanisms, and Models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef]
- Shibata, T.; Ling, F. DNA Recombination Protein-Dependent Mechanism of Homoplasmy and Its Proposed Functions. Mitochondrion 2007, 7, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hunt, C.P.; Rooney, J.P.; Ryde, I.T.; Anbalagan, C.; Joglekar, R.; Meyer, J.N. PCR-Based Analysis of Mitochondrial DNA Copy Number, Mitochondrial DNA Damage, and Nuclear DNA Damage. Curr. Protoc. Toxicol. 2016, 67, 20.11. 1–20.11. 25. [Google Scholar] [CrossRef] [PubMed]
- Baruffini, E.; Ferrari, J.; Dallabona, C.; Donnini, C.; Lodi, T. Polymorphisms in DNA Polymerase γ Affect the MtDNA Stability and the NRTI-Induced Mitochondrial Toxicity in Saccharomyces cerevisiae. Mitochondrion 2015, 20, 52–63. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 12, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, D.; Brunner, M.; Neupert, W.; Westermann, B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 32, 20150–20155. [Google Scholar] [CrossRef]
- Zick, M.; Duvezin-Caubet, S.; Schäfer, A.; Vogel, F.; Neupert, W.; Reichert, A.S. Distinct roles of the two isoforms of the dynamin-like GTPase Mgm1 in mitochondrial fusion. FEBS Lett. 2009, 13, 2237–2243. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–889. [Google Scholar] [CrossRef]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 7844, 57–66. [Google Scholar] [CrossRef]
- Skowronek, E.; Grzechnik, P.; Späth, B.; Marchfelder, A.; Kufel, J. TRNA 3’ Processing in Yeast Involves TRNase Z, Rex1, and Rrp6. RNA 2014, 20, 115–130. [Google Scholar] [CrossRef]
- Montanari, A.; Besagni, C.; De Luca, C.; Morea, V.; Oliva, R.; Tramontano, A.; Bolotin-Fukuhara, M.; Frontali, L.; Francisci, S. Yeast as a Model of Human Mitochondrial TRNA Base Substitutions: Investigation of the Molecular Basis of Respiratory Defects. RNA 2008, 14, 275–283. [Google Scholar] [CrossRef]
- Funes, S.; Herrmann, J.M. Analysis of Mitochondrial Protein Synthesis in Yeast. Methods Mol. Biol. 2007, 372, 255–263. [Google Scholar] [CrossRef]
- Marres, C.A.; de Vries, S.; Grivell, L.A. Isolation and Inactivation of the Nuclear Gene Encoding the Rotenone-Insensitive Internal NADH: Ubiquinone Oxidoreductase of Mitochondria from Saccharomyces cerevisiae. Eur. J. Biochem. 1991, 195, 857–862. [Google Scholar] [CrossRef]
- Schägger, H.; von Jagow, G. Blue Native Electrophoresis for Isolation of Membrane Protein Complexes in Enzymatically Active Form. Anal. Biochem. 1991, 199, 223–231. [Google Scholar] [CrossRef]
- Nijtmans, L.G.J.; Henderson, N.S.; Holt, I.J. Blue Native Electrophoresis to Study Mitochondrial and Other Protein Complexes. Methods 2002, 26, 327–334. [Google Scholar] [CrossRef]
- Jha, P.; Wang, X.; Auwerx, J. Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Dallabona, C.; Marsano, R.M.; Arzuffi, P.; Ghezzi, D.; Mancini, P.; Zeviani, M.; Ferrero, I.; Donnini, C. Sym1, the Yeast Ortholog of the MPV17 Human Disease Protein, Is a Stress-Induced Bioenergetic and Morphogenetic Mitochondrial Modulator. Hum. Mol. Genet. 2010, 19, 1098–1107. [Google Scholar] [CrossRef]
- Pir, P.; Gutteridge, A.; Wu, J.; Rash, B.; Kell, D.B.; Zhang, N.; Oliver, S.G. The Genetic Control of Growth Rate: A Systems Biology Study in Yeast. BMC Syst. Biol. 2012, 6, 4. [Google Scholar] [CrossRef]
- Deutschbauer, A.M.; Jaramillo, D.F.; Proctor, M.; Kumm, J.; Hillenmeyer, M.E.; Davis, R.W.; Nislow, C.; Giaever, G. Mechanisms of Haploinsufficiency Revealed by Genome-Wide Profiling in Yeast. Genetics 2005, 169, 1915–1925. [Google Scholar] [CrossRef]
- Le Roux, B.; Lenaers, G.; Zanlonghi, X.; Amati-Bonneau, P.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 Unique Variants and 831 Patients Registered in an Updated Centralized Variome Database. Orphanet J. Rare Dis. 2019, 14, 214. [Google Scholar] [CrossRef]
- Rahman, S.; Copeland, W.C. POLG-Related Disorders and Their Neurological Manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. Strategies for Fighting Mitochondrial Diseases. J. Intern. Med. 2020, 287, 665–684. [Google Scholar] [CrossRef]
- Rötig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and Mitochondrial Iron-Sulphur Protein Deficiency in Friedreich Ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef]
- Cotticelli, M.G.; Rasmussen, L.; Kushner, N.L.; McKellip, S.; Sosa, M.I.; Manouvakhova, A.; Feng, S.; White, E.L.; Maddry, J.A.; Heemskerk, J.; et al. Primary and Secondary Drug Screening Assays for Friedreich Ataxia. J. Biomol. Screen 2012, 17, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Couplan, E.; Aiyar, R.S.; Kucharczyk, R.; Kabala, A.; Ezkurdia, N.; Gagneur, J.; St Onge, R.P.; Salin, B.; Soubigou, F.; Le Cann, M.; et al. A Yeast-Based Assay Identifies Drugs Active against Human Mitochondrial Disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 11989–11994. [Google Scholar] [CrossRef] [PubMed]
- Hoon, S.; St Onge, R.P.; Giaever, G.; Nislow, C. Yeast Chemical Genomics and Drug Discovery: An Update. Trends Pharmacol. Sci. 2008, 29, 499–504. [Google Scholar] [CrossRef]
- Aiyar, R.S.; Bohnert, M.; Duvezin-Caubet, S.; Voisset, C.; Gagneur, J.; Fritsch, E.S.; Couplan, E.; von der Malsburg, K.; Funaya, C.; Soubigou, F.; et al. Mitochondrial Protein Sorting as a Therapeutic Target for ATP Synthase Disorders. Nat. Commun. 2014, 5, 5585. [Google Scholar] [CrossRef]
- Panozzo, C.; Laleve, A.; Tribouillard-Tanvier, D.; Ostojić, J.; Sellem, C.H.; Friocourt, G.; Bourand-Plantefol, A.; Burg, A.; Delahodde, A.; Blondel, M.; et al. Chemicals or Mutations That Target Mitochondrial Translation Can Rescue the Respiratory Deficiency of Yeast Bcs1 Mutants. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2297–2307. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, Is Mutated in Dominant Optic Atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, Encoding a Dynamin-Related GTPase, Is Mutated in Autosomal Dominant Optic Atrophy Linked to Chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Amati-Bonneau, P.; Milea, D.; Bonneau, D.; Chevrollier, A.; Ferré, M.; Guillet, V.; Gueguen, N.; Loiseau, D.; de Crescenzo, M.-A.P.; Verny, C.; et al. OPA1-Associated Disorders: Phenotypes and Pathophysiology. Int. J. Biochem. Cell Biol. 2009, 41, 1855–1865. [Google Scholar] [CrossRef]
- Jones, B.A.; Fangman, W.L. Mitochondrial DNA Maintenance in Yeast Requires a Protein Containing a Region Related to the GTP-Binding Domain of Dynamin. Genes Dev. 1992, 6, 380–389. [Google Scholar] [CrossRef]
- Aleo, S.J.; Del Dotto, V.; Fogazza, M.; Maresca, A.; Lodi, T.; Goffrini, P.; Ghelli, A.; Rugolo, M.; Carelli, V.; Baruffini, E.; et al. Drug Repositioning as a Therapeutic Strategy for Neurodegenerations Associated with OPA1 Mutations. Hum. Mol. Genet. 2021, 29, 3631–3645. [Google Scholar] [CrossRef] [PubMed]
- Pitayu, L.; Baruffini, E.; Rodier, C.; Rötig, A.; Lodi, T.; Delahodde, A. Combined Use of Saccharomyces cerevisiae, Caenorhabditis Elegans and Patient Fibroblasts Leads to the Identification of Clofilium Tosylate as a Potential Therapeutic Chemical against POLG-Related Diseases. Hum. Mol. Genet. 2016, 25, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Facchinello, N.; Laquatra, C.; Locatello, L.; Beffagna, G.; Brañas Casas, R.; Fornetto, C.; Dinarello, A.; Martorano, L.; Vettori, A.; Risato, G.; et al. Efficient Clofilium Tosylate-Mediated Rescue of POLG-Related Disease Phenotypes in Zebrafish. Cell Death Dis. 2021, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Tigano, M.; Ruotolo, R.; Dallabona, C.; Fontanesi, F.; Barrientos, A.; Donnini, C.; Ottonello, S. Elongator-Dependent Modification of Cytoplasmic TRNALysUUU Is Required for Mitochondrial Function under Stress Conditions. Nucleic Acids Res. 2015, 43, 8368–8380. [Google Scholar] [CrossRef]
- Varghese, F.; Atcheson, E.; Bridges, H.R.; Hirst, J. Characterization of clinically identified mutations in NDUFV1, the flavin-binding subunit of respiratory complex I, using a yeast model system. Hum. Mol. Genet. 2015, 22, 6350–6360. [Google Scholar] [CrossRef]


{kind=link}
| Function | Human/Yeast Gene | References * | |
|---|---|---|---|
| OXPHOS subunits | CII | SDHA/SDH1 | [38] |
| SDHB/SDH2 | [39,40] | ||
| SDHD/SDH4 | [41,42] | ||
| CIV | COX6B1/COX12 | [43] | |
| CV | ATP5E/ATP15 | [44] | |
| OXPHOS assembly factors | CII | SDHAF1/SDH6 | [45] |
| CIII | BCS1L/BCS1 | [46,47,48,49,50] | |
| LYRM7/MZM1 | [51,52] | ||
| CIV | COX10/COX10 | [53,54] | |
| SURF1/SHY1 | [55,56] | ||
| CV | ATPAF2/ATP12 | [57,58] | |
| Protein import and processing | AFG3L2/AFG3 | [59,60,61,62,63,64] | |
| GFER/ERV1 | [65,66] | ||
| MIPEP/OCT1 | [67] | ||
| PITRM1/CYM1 | [68,69] | ||
| PMPCB/MAS1 | [70] | ||
| SPG7/YTA12 | [60,62,63] | ||
| TIMM50/TIM50 | [71] | ||
| TIMM8A/TIM8 | [72,73] | ||
| mtDNA replication, transcription and maintenance | MPV17/SYM1 | [74,75] | |
| POLG/MIP1 | [76,77,78,79,80,81,82,83,84,85,86,87,88,89,90] | ||
| POLR2A/RPB1 | [91] | ||
| RNA maturation/modification | ELAC2/TRZ1 | [92] | |
| GTPBP3/MSS1 | [93] | ||
| MRM2/MRM2 | [94] | ||
| MTO1/MTO1 | [95,96] | ||
| TRIT1/MOD5 | [97] | ||
| TRMT5/TRM5 | [98] | ||
| TRMU/MTO2 | [99,100] | ||
| TRNT1/CCA1 | [101,102] | ||
| Mitochondrial aminoacyl tRNA synthetases | AARS2/ALA1 | [103,104] | |
| GARS/GRS1 | [105,106,107] | ||
| GATB/PET112 | [108] | ||
| HARS2/HTS1 | [109] | ||
| KARS/MSK1 | [110] | ||
| LARS2/NAM2 | [111] | ||
| QRSL1/HERS2 | [108] | ||
| RARS2/MSR1 | [112] | ||
| TARS2/MST1-THS1 | [113] | ||
| VARS2/VAS1 | [114,115] | ||
| WARS2/MSW1 | [116] | ||
| YARS2/MSY1 | [117,118,119] | ||
| Translation | GFM1/MEF1 | [120] | |
| TUFM/TUF1 | [120,121,122] | ||
| Membrane dynamics and composition | DNM1L/DNM1 | [123,124,125] | |
| MFN2/FZO1 | [126] | ||
| OPA1/MGM1 | [127,128] | ||
| TAZ/TAZ1 | [129,130,131] | ||
| VPS13C/VPS13 | [132,133,134] | ||
| APOO/MIC26 paralog MIC27 | [135] | ||
| GDAP1/ | [136] | ||
| ACO2/ACO1 | [137,138,139,140] | ||
| IDH3A/IDH2 | [141] | ||
| MDH2/MDH1 | [142] | ||
| MECR/ETR1 | [143] | ||
| MPC1/MPC1 | [144] | ||
| PDHA1/PDA1 | [145] | ||
| PDHX/PDX1 | [146] | ||
| PPA2/PPA2 | [147] | ||
| SLC25A13/AGC1 | [148] | ||
| SLC25A3/PIC2-MIR1 | [149,150] | ||
| Fe–S cluster biogenesis | ABCB7/ATM1 | [151,152,153] | |
| FDXR/ARH1 | [154] | ||
| FXN/YFH1 | [155,156,157,158,159,160] | ||
| ISCU/ISU1 paralog ISU2 | [161,162] | ||
| LYRM4/ISD11 | [163] | ||
| NFU1/NFU1 | [164] | ||
| Enzyme co-factors | COASY/CAB5 | [165,166] | |
| LIPT1/LIP3 | [167] | ||
| LIPT2/LIP2 | [168] | ||
| PANK2/CAB1 | [169] | ||
| Metabolite transport | SLC25A1/CTP1 | [170] | |
| SLC25A4/AAC2 | [171,172,173,174,175,176,177,178,179] | ||
| SLC25A19/TPC1 | [180] | ||
| SLC25A32/FLX1 | [181] | ||
| Electron carriers | CoQ | COA6/COA6 | [182,183] |
| COQ2/COQ2 | [184,185,186,187] | ||
| COQ4/COQ4 | [188,189] | ||
| COQ5/COQ5 | [190] | ||
| COQ6/COQ6 | [191,192] | ||
| COQ8A/COQ8 | [193] | ||
| COQ8B/COQ8 | [194] | ||
| COQ9/COQ9 | [195,196] | ||
| PDSS1/COQ1 | [187] | ||
| Cytc | CYCS/CYC1 paralog CYC7 | [197,198] | |
| HCCS/CYC3 | [199,200,201] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccatelli Berti, C.; di Punzio, G.; Dallabona, C.; Baruffini, E.; Goffrini, P.; Lodi, T.; Donnini, C. The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases. Genes 2021, 12, 300. https://doi.org/10.3390/genes12020300
Ceccatelli Berti C, di Punzio G, Dallabona C, Baruffini E, Goffrini P, Lodi T, Donnini C. The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases. Genes. 2021; 12(2):300. https://doi.org/10.3390/genes12020300
Chicago/Turabian StyleCeccatelli Berti, Camilla, Giulia di Punzio, Cristina Dallabona, Enrico Baruffini, Paola Goffrini, Tiziana Lodi, and Claudia Donnini. 2021. "The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases" Genes 12, no. 2: 300. https://doi.org/10.3390/genes12020300
APA StyleCeccatelli Berti, C., di Punzio, G., Dallabona, C., Baruffini, E., Goffrini, P., Lodi, T., & Donnini, C. (2021). The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases. Genes, 12(2), 300. https://doi.org/10.3390/genes12020300