De Novo Development of mtDNA Deletion Due to Decreased POLG and SSBP1 Expression in Humans


 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. PCR Assay for Large mtDNA Deletions
2.2. Quantitative PCR for the Detection of mtDNA Deletions
2.3. Skin Fibroblast Isolation and Culture, and Single-Cell Sub-cloning
2.4. Mitochondrial Respiration
2.5. Exome Sequencing
2.6. Sanger Sequencing
2.7. Quantitative RT-PCR (qRT-PCR) for POLG and DNMT Expression
2.8. Protein Preparation and Western Blot
2.9. Methylation Analysis by Pyrosequencing
2.10. Statistical Analysis
3. Results
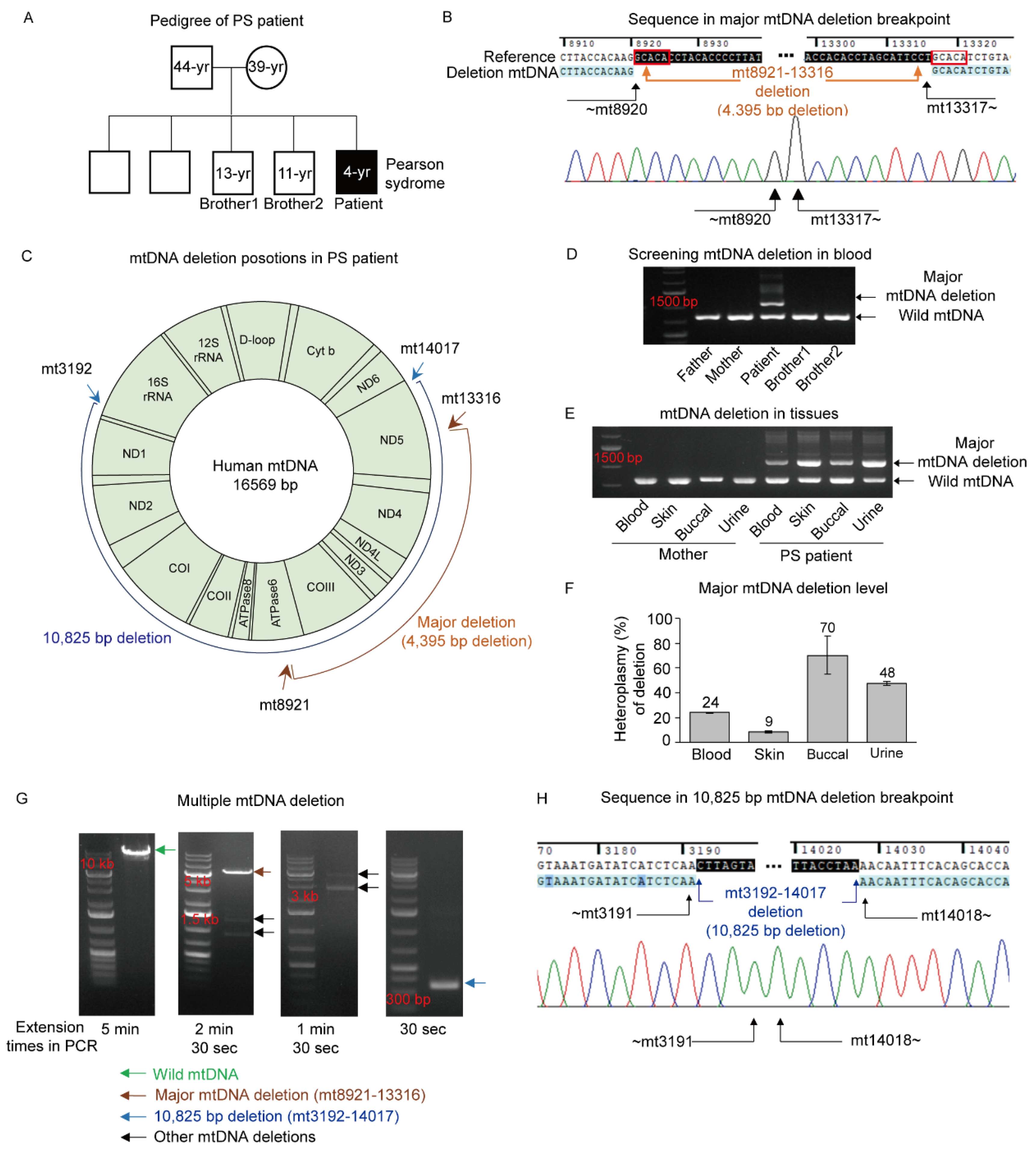
3.1. Pearson Syndrome (PS) with a Large mtDNA Deletion
3.2. Identification of Nuclear POLG and SSBP1 Mutations by Exome Sequencing
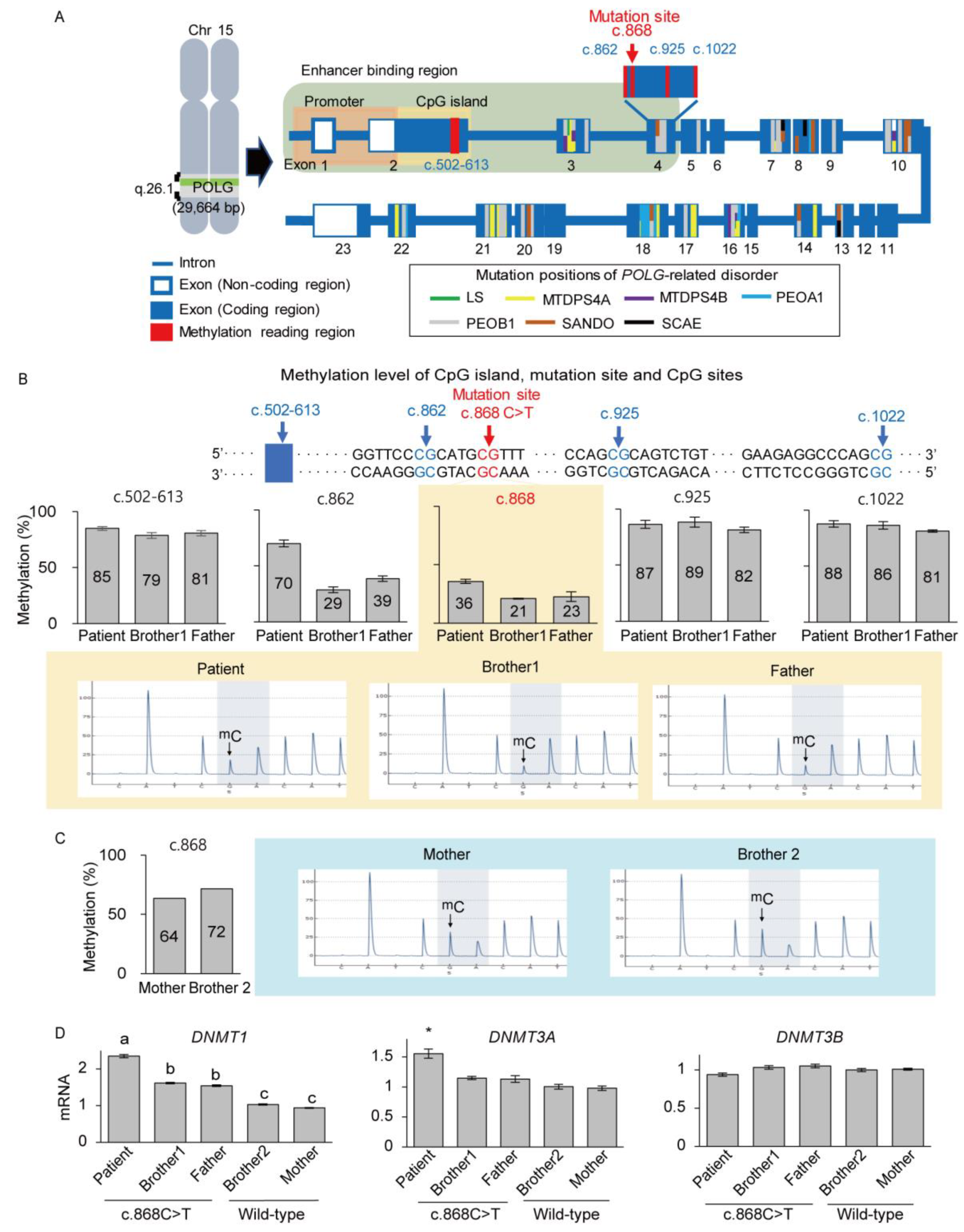
3.3. Deficient Expression of POLG and SSBP1 Induced by Mutations in POLG and SSBP1 Genes
3.4. De Novo mtDNA Deletions with Methylation Changes
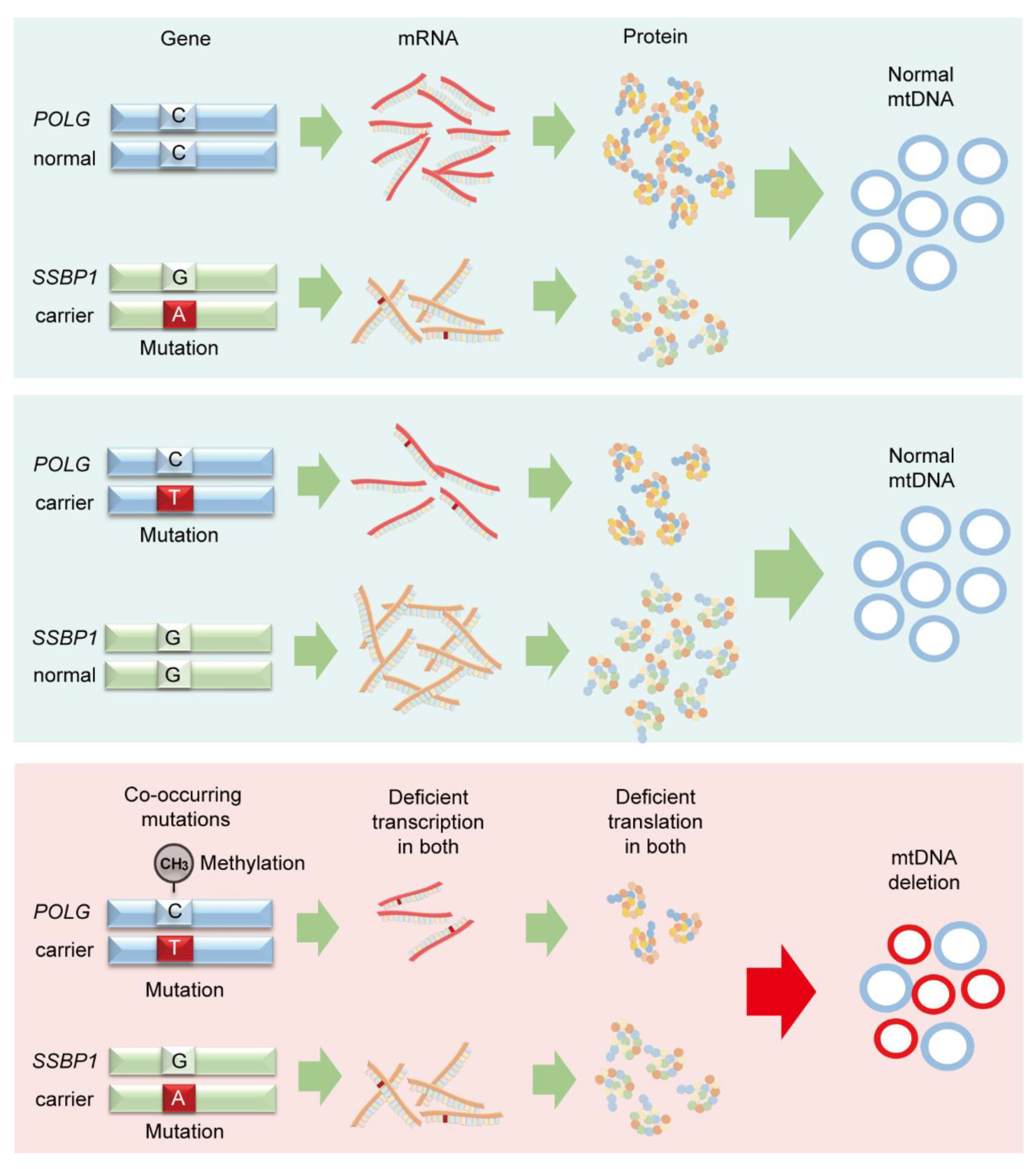
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Filler, K.; Lyon, D.; Bennett, J.; McCain, N.; Elswick, R.; Lukkahatai, N.; Saligan, L.N. Association of Mitochondrial Dysfunction and Fatigue: A Review of the Literature. BBA Clin. 2014, 1, 12–23. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Zapico, S.C.; Ubelaker, D.H. mtDNA Mutations and Their Role in Aging, Diseases and Forensic Sciences. Aging Dis. 2013, 4, 364–380. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef]
- Ferreira, M.; Evangelista, T.; Almeida, L.S.; Martins, J.; Macario, M.C.; Martins, E.; Moleirinho, A.; Azevedo, L.; Vilarinho, L.; Santorelli, F.M. Relative frequency of known causes of multiple mtDNA deletions: Two novel POLG mutations. Neuromuscul. Disord. 2011, 21, 483–488. [Google Scholar] [CrossRef]
- Ro, L.S.; Lai, S.L.; Chen, C.M.; Chen, S.T. Deleted 4977-bp mitochondrial DNA mutation is associated with sporadic amyotrophic lateral sclerosis: A hospital-based case-control study. Muscle Nerve 2003, 28, 737–743. [Google Scholar] [CrossRef]
- Meissner, C.; Bruse, P.; Mohamed, S.A.; Schulz, A.; Warnk, H.; Storm, T.; Oehmichen, M. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: A useful biomarker or more? Exp. Gerontol. 2008, 43, 645–652. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef]
- Schon, E.A.; Rizzuto, R.; Moraes, C.T.; Nakase, H.; Zeviani, M.; DiMauro, S. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science 1989, 244, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Knoll, N.; Jarick, I.; Volckmar, A.L.; Klingenspor, M.; Illig, T.; Grallert, H.; Gieger, C.; Wichmann, H.E.; Peters, A.; Hebebrand, J.; et al. Gene set of nuclear-encoded mitochondrial regulators is enriched for common inherited variation in obesity. PLoS ONE 2013, 8, e55884. [Google Scholar] [CrossRef]
- Pinz, K.G.; Bogenhagen, D.F. Characterization of a catalytically slow AP lyase activity in DNA polymerase gamma and other family A DNA polymerases. J. Biol. Chem. 2000, 275, 12509–12514. [Google Scholar] [CrossRef]
- Hudson, G.; Chinnery, P.F. Mitochondrial DNA polymerase-gamma and human disease. Hum. Mol. Genet. 2006, 15, 244. [Google Scholar] [CrossRef]
- Hanisch, F.; Kornhuber, M.; Alston, C.L.; Taylor, R.W.; Deschauer, M.; Zierz, S. SANDO syndrome in a cohort of 107 patients with CPEO and mitochondrial DNA deletions. J. Neurol. Neurosurg. Psychiatry. 2015, 86, 630–634. [Google Scholar] [CrossRef]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef]
- Miralles Fusté, J.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, Ö.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In vivo occupancy of mitochondrial single-stranded DNA binding protein supports the strand displacement mode of DNA. PLoS Genet. 2014, 10, e1004832. [Google Scholar] [CrossRef]
- Del Dotto, V.; Ullah, F.; Di Meo, I.; Magini, P.; Gusic, M.; Maresca, A.; Caporali, L.; Palombo, F.; Tagliavini, F.; Baugh, E.H.; et al. SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. J. Clin. Investig. 2020, 130, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Wang, X.; Tippner-Hedges, R.; Ma, H.; Folmes, C.D.; Gutierrez, N.M.; Lee, Y.; Van Dyken, C.; Ahmed, R.; Li, Y.; et al. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs. Cell. Stem Cell. 2016, 18, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Okae, H.; Toh, H.; Chiba, H.; Hiura, H.; Shirane, K.; Sato, T.; Suyama, M.; Yaegashi, N.; Sasaki, H.; et al. Allele-Specific Methylome and Transcriptome Analysis Reveals Widespread Imprinting in the Human Placenta. Am. J. Hum. Genet. 2016, 99, 1045–1058. [Google Scholar] [CrossRef]
- Ferreira, I.D.; Rosario, V.E.; Cravo, P.V. Real-time quantitative PCR with SYBR Green I detection for estimating copy numbers of nine drug resistance candidate genes in Plasmodium falciparum. Malar. J. 2006, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.H.; Kang, E.; Kim, Y.M.; Go, H.; Kim, K.Y.; Jung, J.Y.; Kang, M.; Kim, G.H.; Kim, J.M.; Choi, I.H.; et al. Fabry disease: Characterization of the plasma proteome pre- and post-enzyme replacement therapy. J. Med. Genet. 2017, 54, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Madi, T.; Balamurugan, K.; Bombardi, R.; Duncan, G.; McCord, B. The determination of tissue-specific DNA methylation patterns in forensic biofluids using bisulfite modification and pyrosequencing. Electrophoresis 2012, 33, 1736–1745. [Google Scholar] [CrossRef]
- Ma, H.; Folmes, C.D.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 2015, 524, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, X.; Bin, R.; Yu, S.; Xia, Z.; Zheng, G.; Zhong, J.; Zhang, Y.; Jiang, Y.H.; Wang, Y. Genetic Variants Identified from Epilepsy of Unknown Etiology in Chinese Children by Targeted Exome Sequencing. Sci. Rep. 2017, 7, 40319. [Google Scholar] [CrossRef] [PubMed]
- Jurkute, N.; Leu, C.; Pogoda, H.M.; Arno, G.; Robson, A.G.; Nürnberg, G.; Altmüller, J.; Thiele, H.; Motameny, S.; Toliat, M.R.; et al. SSBP1 mutations in dominant optic atrophy with variable retinal degeneration. Ann. Neurol. 2019, 86, 368–383. [Google Scholar] [CrossRef]
- Piro-Mégy, C.; Sarzi, E.; Tarrés-Solé, A.; Péquignot, M.; Hensen, F.; Quilès, M.; Manes, G.; Chakraborty, A.; Sénéchal, A.; Bocquet, B.; et al. Dominant mutations in mtDNA maintenance gene SSBP1 cause optic atrophy and foveopathy. J. Clin. Investig. 2020, 2, 143–156. [Google Scholar]
- Gustafson, M.A.; McCormick, E.M.; Perera, L.; Longley, M.J.; Bai, R.; Kong, J.; Dulik, M.; Shen, L.; Goldstein, A.C.; McCormack, S.E.; et al. Mitochondrial single-stranded DNA binding protein novel de novo SSBP1 mutation in a child with single large-scale mtDNA deletion (SLSMD) clinically manifesting as Pearson, Kearns-Sayre, and Leigh syndromes. PLoS ONE 2019, 14, e0221829. [Google Scholar] [CrossRef]
- Lindgren, U.; Roos, S.; Hedberg Oldfors, C.; Moslemi, A.R.; Lindberg, C.; Oldfors, A. Mitochondrial pathology in inclusion body myositis. Neuromuscul. Disord. 2015, 25, 281–288. [Google Scholar] [CrossRef]
- Longley, M.J.; Clark, S.; Yu Wai Man, C.; Hudson, G.; Durham, S.E.; Taylor, R.W.; Nightingale, S.; Turnbull, D.M.; Copeland, W.C.; Chinnery, P.F. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am. J. Hum. Genet. 2006, 78, 1026–1034. [Google Scholar] [CrossRef]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef] [PubMed]
- Samuels, D.C.; Schon, E.A.; Chinnery, P.F. Two direct repeats cause most human mtDNA deletions. Trends Genet. 2004, 20, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Löfgren, A.; Dermaut, B.; Ceuterick, C.; Martin, J.J.; Van Broeckhoven, C. Digenic progressive external ophthalmoplegia in a sporadic patient: Recessive mutations in POLG and C10orf2/Twinkle. Hum. Mutat. 2003, 22, 175–176. [Google Scholar] [CrossRef] [PubMed]
- King, A.D.; Huang, K.; Rubbi, L.; Liu, S.; Wang, C.Y.; Wang, Y.; Pellegrini, M.; Fan, G. Reversible Regulation of Promoter and Enhancer Histone Landscape by DNA Methylation in Mouse Embryonic Stem Cells. Cell. Rep. 2016, 17, 289–302. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Clinical Features |
|---|---|
| 5 months | Sideroblastic anemia Pancytopenia |
| Bone marrow failure | |
| 2 years | Proximal renal tubular acidosis |
| 3 years | Developmental delay |
| Exocrine pancreatic insufficiency | |
| Adrenal cortical insufficiency | |
| Chronic kidney disease |
| Mutation in POLG (Allele/Allele) | POLG Exon | mtDNA Deletion | Disease/Clinical Features | Reported Year | Reference |
|---|---|---|---|---|---|
| p.A467T/p.W748S | 7/13 | Multiple | Sensory ataxic neuropathy with dysarthria and ophthalmoparesis (SANDO) | 2018 | [14] |
| p.P587L + p.T251I/p.R869Q | 10+3/17 | Multiple | 2018 | [14] | |
| p.A467Thr/p.A467T | 7/7 | Multiple | 2018 | [14] | |
| p.A467Thr/p.R627Q | 7/10 | Multiple | 2018 | [14] | |
| p.P648R/p.R807C | 10/10 | Multiple | 2011 | [6] | |
| p.Q1236H/wt | 23 | Single | Inclusion body myositis (IBM) | 2015 | [28] |
| p.G451E/wt | 7 | Multiple | Progressive external ophthalmoplegia (PEO) | 2006 | [29] |
| p.M430L/p.W918R | 7/18 | Multiple | 2011 | [6] | |
| p.G517V/wt | 8 | Multiple | 2011 | [6] | |
| p.W585X/p.P648R | 10/10 | Multiple | 2011 | [6] | |
| p.A862T/p.R1081Q | 16/20 | Multiple | Alpers-like | 2011 | [6] |
| p.R1081Q/wt | 20 | Multiple | Muscle weakness, ataxia, extrapyramidal features, diabetes mellitus | 2011 | [6] |
| p.R807C/wt | 14 | Multiple | Proximal myopathy, ptosis, diplopia | 2011 | [6] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.; Kim, T.; Lee, M.; So, S.; Karagozlu, M.Z.; Seo, G.H.; Choi, I.H.; Lee, P.C.W.; Kim, C.-J.; Kang, E.; et al. De Novo Development of mtDNA Deletion Due to Decreased POLG and SSBP1 Expression in Humans. Genes 2021, 12, 284. https://doi.org/10.3390/genes12020284
Lee Y, Kim T, Lee M, So S, Karagozlu MZ, Seo GH, Choi IH, Lee PCW, Kim C-J, Kang E, et al. De Novo Development of mtDNA Deletion Due to Decreased POLG and SSBP1 Expression in Humans. Genes. 2021; 12(2):284. https://doi.org/10.3390/genes12020284
Chicago/Turabian StyleLee, Yeonmi, Taeho Kim, Miju Lee, Seongjun So, Mustafa Zafer Karagozlu, Go Hun Seo, In Hee Choi, Peter C. W. Lee, Chong-Jai Kim, Eunju Kang, and et al. 2021. "De Novo Development of mtDNA Deletion Due to Decreased POLG and SSBP1 Expression in Humans" Genes 12, no. 2: 284. https://doi.org/10.3390/genes12020284
APA StyleLee, Y., Kim, T., Lee, M., So, S., Karagozlu, M. Z., Seo, G. H., Choi, I. H., Lee, P. C. W., Kim, C.-J., Kang, E., & Lee, B. H. (2021). De Novo Development of mtDNA Deletion Due to Decreased POLG and SSBP1 Expression in Humans. Genes, 12(2), 284. https://doi.org/10.3390/genes12020284