
Prediction Accuracies of Genomic Selection for Nine Commercially Important Traits in the Portuguese Oyster (Crassostrea angulata) Using DArT-Seq Technology


 ,
, 
Abstract
1. Introduction
2. Material and Methods
2.1. Oyster Provenance
2.2. Phenotypic Measurements
2.3. DNA Extraction, Library Construction, SNP Genotyping and Quality Control
2.4. Genetic Parameter Estimation
2.5. Genomic Prediction Accuracy
2.6. Cross Validation
3. Results and Discussion
3.1. Trait Summary and Genetic Parameter Estimates
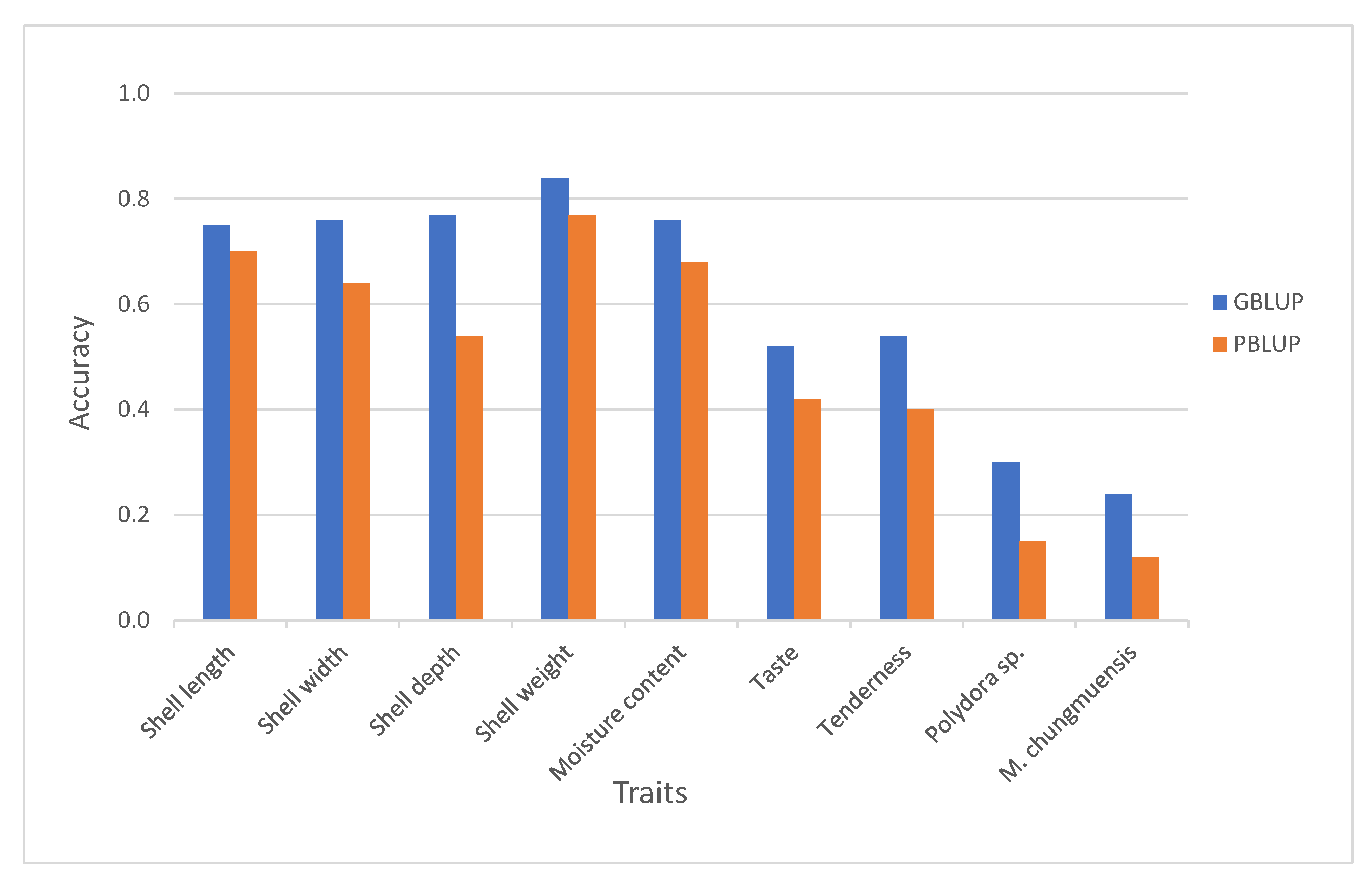
3.2. Reliability of Different Genomic Selection Methods
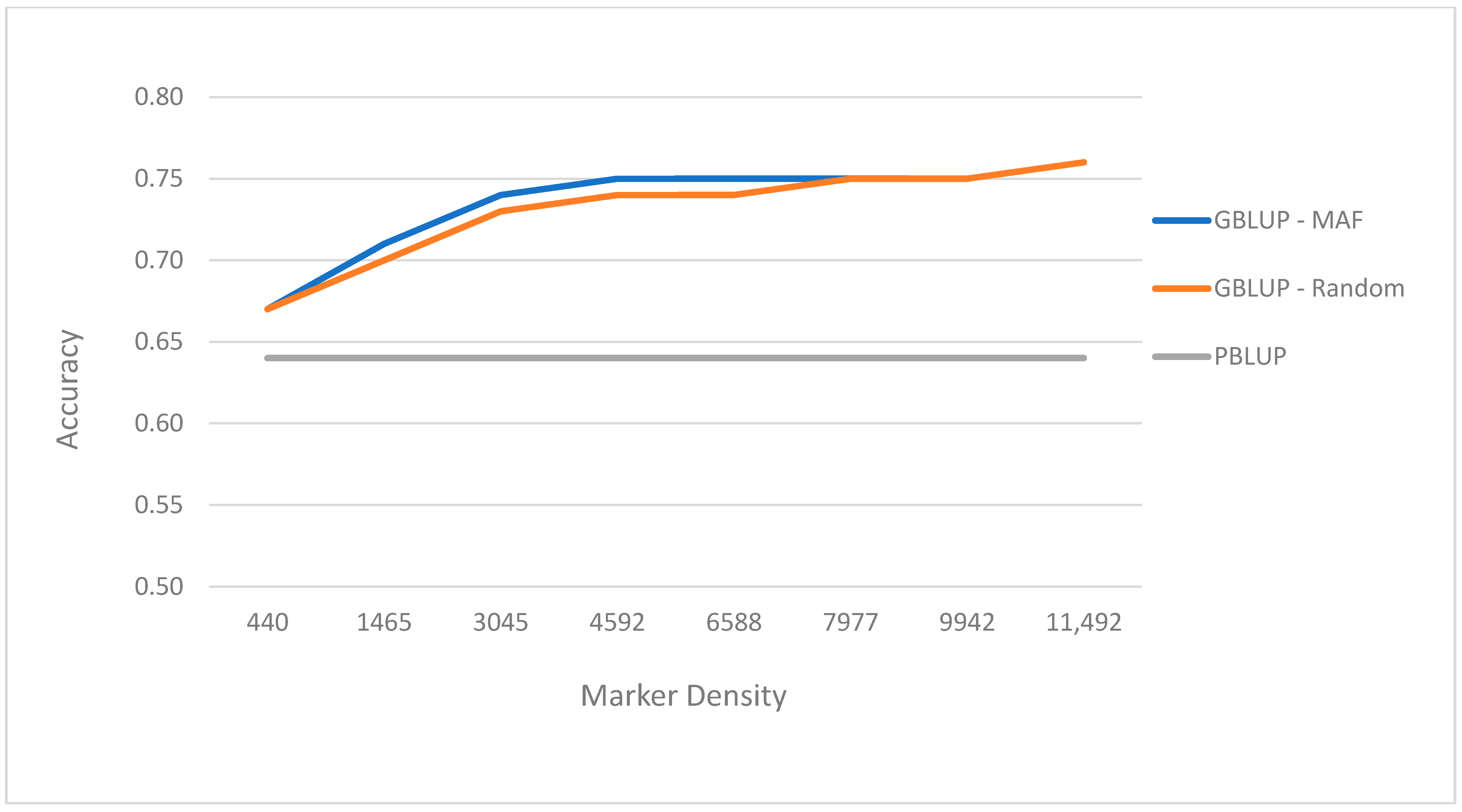
3.3. Prediction Performance with Different Number of SNP Sets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gagnaire, P.-A.; Lamy, J.-B.; Cornette, F.; Heurtebise, S.; Dégremont, L.; Flahauw, E.; Boudry, P.; Bierne, N.; Lapègue, S. Analysis of Genome-Wide Differentiation between Native and Introduced Populations of the Cupped Oysters Crassostrea gigas and Crassostrea angulata. Genome Biol. Evol. 2018, 10, 2518–2534. [Google Scholar] [CrossRef] [PubMed]
- Grade, A.; Chairi, H.; Lallias, D.; Power, D.M.; Ruano, F.; Leitão, A.; Drago, T.; King, J.W.; Boudry, P.; Batista, F.M. New insights about the introduction of the Portuguese oyster, Crassostrea angulata, into the North East Atlantic from Asia based on a highly polymorphic mitochondrial region. Aquat. Living Resour. 2016, 29, 404. [Google Scholar] [CrossRef]
- Vu, V.I.; O’Connor, W.; Vu, V.S.; Phan, T.V.; Knibb, W. Resolution of the controversial relationship between Pacific and Portuguese oysters internationally and in Vietnam. Aquaculture 2017, 473, 389–399. [Google Scholar]
- Bureau, C.F. China Fisheries Yearbook; Chinese Agriculture Express: Beijing, China, 2013; pp. 23–52. [Google Scholar]
- O’Connor, W.; Dove, M.; O’Connor, S.; Van In, V.; Lien MV, T.N.; Van, P.T. Project Enhancing Bivalve Production in Northern Vietnam & NSW; ACIAR: Canberra, Australia, 2019.
- Mizuta, D.D.; Wikfors, G.H. Seeking the perfect oyster shell: A brief review of current knowledge. Rev. Aquac. 2018, 11, 586–602. [Google Scholar] [CrossRef]
- Marshall, R.; Dunham, A. Effects of culture media and stocking density on biofouling, shell shape, growth, and survival of the Pacific oyster (Crassostrea gigas) and the Manila clam (Venerupis philippinarum) in suspended culture. Aquaculture 2013, 406, 68–78. [Google Scholar] [CrossRef]
- Gosling, E. Bivalve Molluscs: Biology, Ecology and Culture. In Fishing New Books; Blackwell Science: Oxford, UK, 2004. [Google Scholar]
- Vu, S.V.; Knibb, W.; Nguyen, N.T.; Vu, I.V.; O’Connor, W.; Dove, M.; Nguyen, N.H. First breeding program of the Portuguese oyster Crassostrea angulata demonstrated significant selection response in traits of economic importance. Aquaculture 2020, 518, 734664. [Google Scholar] [CrossRef]
- Vu, S.V.; Knibb, W.; O’Connor, W.; Nguyen, N.T.; Van In, V.; Dove, M.; Nguyen, N.H. Genetic parameters for traits affecting consumer preferences for the Portuguese oyster, Crassostrea angulata. Aquaculture 2020, 526, 735391. [Google Scholar] [CrossRef]
- Goddard, M.E.; Hayes, B.J. Genomic selection. J. Anim. Breed. Genet. 2007, 124, 323–330. [Google Scholar] [CrossRef]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Meuwissen, T.; Hayes, B.; Goddard, M. Accelerating Improvement of Livestock with Genomic Selection. Annu. Rev. Anim. Biosci. 2013, 1, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Premachandra, H.; Kilian, A.; Knibb, W. Genomic prediction using DArT-Seq technology for yellowtail kingfish Seriola lalandi. BMC Genom. 2018, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Phuthaworn, C.; Knibb, W. Genomic prediction for disease resistance to Hepatopancreatic parvovirus and growth, carcass and quality traits in Banana shrimp Fenneropenaeus merguiensis. Genomics 2020, 112, 2021–2027. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Dong, L.; Gu, L.; Han, Z.; Zhang, W.; Fang, M.; Wang, Z. Evaluation of Genomic Selection for Seven Economic Traits in Yellow Drum (Nibea albiflora). Mar. Biotechnol. 2019, 21, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.M.; Lhorente, J.P.; Correa, K.; Soto, J.; Salas, D.; Yáñez, J.M. Genome-wide association study and cost-efficient genomic predictions for growth and fillet yield in Nile tilapia (Oreochromis niloticus). G3 Genes Genomes Genet. 2019, 9, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Skaarud, A.; De Vera, M.; Alvarez, A.; Ødegård, J. Genomic prediction for commercial traits using univariate and multivariate approaches in Nile tilapia (Oreochromis niloticus). Aquaculture 2020, 516, 734641. [Google Scholar] [CrossRef]
- Castillo-Juárez, H.; Campos-Montes, G.R.; Caballero-Zamora, A.; Montaldo, H.H. Genetic improvement of Pacific white shrimp [Penaeus (Litopenaeus) vannamei]: Perspectives for genomic selection. Front. Genet. 2015, 6, 93. [Google Scholar] [CrossRef]
- Zenger, K.R.; Khatkar, M.S.; Jones, D.B.; Khalilisamani, N.; Jerry, D.R.; Raadsma, H.W. Genomic Selection in Aquaculture: Application, Limitations and Opportunities with Special Reference to Marine Shrimp and Pearl Oysters. Front. Genet. 2019, 9, 693. [Google Scholar] [CrossRef]
- Gutierrez, A.P.; Matika, O.; Bean, T.P.; Houston, R.D. Genomic Selection for Growth Traits in Pacific Oyster (Crassostrea gigas): Potential of Low-Density Marker Panels for Breeding Value Prediction. Front. Genet. 2018, 9, 391. [Google Scholar] [CrossRef]
- Gutierrez, A.P.; Symonds, J.; King, N.; Steiner, K.; Bean, T.P.; Houston, R.D. Potential of genomic selection for improvement of resistance to ostreid herpesvirus in Pacific oyster (Crassostrea gigas). Anim. Genet. 2020, 51, 249–257. [Google Scholar] [CrossRef]
- Jones, D.; Toole, P.; Khatkar, M.; Raadsma, H.; Jerry, D.; Zenger, K. Developing a genomic selection breeding program for complex pearl colour traits within the silver-lipped pearl oyster. Proc. Assoc. Adv. Anim. Breed. Genet. 2017, 22, 537–540. [Google Scholar]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C. Diversity arrays technology: A generic genome profiling technology on open platforms. In Data Production and Analysis in Population Genomics; Springer: New York City, NY, USA, 2012; pp. 67–89. [Google Scholar]
- Vu, V.I.; Vu, V.S.; O’Connor, W.; Phan, T.V.; Dove, M.; Knibb, W.; Nguyen, N.H. Are strain genetic effect and heterosis expression altered with culture system and rearing environment in the Portuguese oyster (Crassostrea angulata)? Aquac. Res. 2017, 48, 4058–4069. [Google Scholar]
- Blake, J.; Evans, J. Polydora and related genera (Polychaeta: Spionidae) as borers in mollusk shells and other calcareous substrates. Veliger 1973, 15, 235–249. [Google Scholar]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. In BMC Proceedings; BioMed Central: London, UK, 2011; Volume 5, p. 54. [Google Scholar]
- Courtois, B.; Audebert, A.; Dardou, A.; Roques, S.; Herrera, T.G.; Droc, G.; Frouin, J.; Rouan, L.; Gozé, E.; Kilian, A.; et al. Genome-Wide Association Mapping of Root Traits in a Japonica Rice Panel. PLoS ONE 2013, 8, e78037. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Pollara, V.J.; Cowles, C.R.; Van Etten, W.J.; Baldwin, J.; Linton, L.; Lander, E.S. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 2000, 407, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- VanRaden, P.; Van Tassell, C.; Wiggans, G.; Sonstegard, T.; Schnabel, R.; Schenkel, F. Reliability of genomic predictions for North American dairy bulls. J. Dairy Sci. 2008, 91, 305. [Google Scholar]
- Mrode, R.A. Linear Models for the Prediction of Animal Breeding Values; CABI: Wallingford, UK, 2014. [Google Scholar]
- Gilmour, A.R.; Thompson, R. ASReml Update. What’s New in Release 4.2; VSM International Ltd.: Hemel Hempstead, UK, 2019. [Google Scholar]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Cheng, H.; Qu, L.; Garrick, D.; Fernando, R.L. A fast and efficient Gibbs sampler for BayesB in whole-genome analyses. Genet. Sel. Evol. 2015, 47, 80. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de los Campos, G. BGLR: A statistical package for whole genome regression and prediction. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, G.; Zeng, Q.; Chen, Z.; Hu, X.; Li, H.; Wang, S.; Bao, Z. Predicting Growth Traits with Genomic Selection Methods in Zhikong Scallop (Chlamys farreri). Mar. Biotechnol. 2018, 20, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Beattie, J.; Chew, K.; Hershberger, W. Differential survival of selected strains of Pacific oysters (Crassostrea gigas) during summer mortality. In Proceedings of the National Shellfisheries Association, San Antonia, TX, USA, December 1980. [Google Scholar]
- Burge, C.; Judah, L.R.; Conquest, L.L.; Griffin, F.J.; Cheney, D.P.; Suhrbier, A.; Vadopalas, B.; Olin, P.G.; Renault, T.; Friedman, C.S. Summer seed mortality of the Pacific oyster, Crassostrea gigas Thunberg grown in Tomales Bay, California, USA: The influence of oyster stock, planting time, pathogens, and environmental stressors. J. Shellfish Res. 2007, 26, 163–172. [Google Scholar] [CrossRef]
- Martin, A.-G.; Gerard, A.; Cochennec, N.; Langlade, A. Selecting flat oysters, Ostrea edulis, for survival against the parasite Bonamia ostreae: Assessment of the resistance of a first selected generation. In Proceedings of the International Conference of Bordeaux Aquaculture ’92, EAS, Bordeaux, France, 25–27 March 1992. [Google Scholar]
- Dove, M.C.; O’Connor, W.; Nell, J.A. Evaluation of the progeny of the fourth-generation Sydney rock oyster Saccostrea glomerata (Gould, 1850) breeding lines for resistance to QX disease (Marteilia sydneyi) and winter mortality (Bonamia roughleyi). Aquac. Res. 2012, 44, 1791–1800. [Google Scholar] [CrossRef]
- Abbe, G.R.; McCollough, C.B.; Barker, L.S.; Dungan, C.F. Performance of Disease-Tolerant Strains of Eastern Oyster (Crassostrea virginica) in the Patuxent River, Maryland, 2003 to 2007. J. Shellfish Res. 2010, 29, 161–175. [Google Scholar] [CrossRef]
- Brake, J.; Evans, F.; Langdon, C. Evidence for genetic control of pigmentation of shell and mantle edge in selected families of Pacific oysters, Crassostrea gigas. Aquaculture 2004, 229, 89–98. [Google Scholar] [CrossRef]
- Falconer, D.S.; Mackay, T.F.C. Introduction to Quantitative Genetics, 4th ed.; Longman: Harlow, UK, 1996. [Google Scholar]
- Ma, P.; Lund, M.; Nielsen, U.; Aamand, G.; Su, G. Single-step genomic model improved reliability and reduced the bias of genomic predictions in Danish Jersey. J. Dairy Sci. 2015, 98, 9026–9034. [Google Scholar] [CrossRef]
- Fernando, R.L.; Cheng, H.; Garrick, D. An efficient exact method to obtain GBLUP and single-step GBLUP when the genomic relationship matrix is singular. Genet. Sel. Evol. 2016, 48, 80. [Google Scholar] [CrossRef]
- Ødegård, J.; Moen, T.; Santi, N.; Korsvoll, S.A.; Kjøglum, S.; Meuwissen, T.H. Genomic prediction in an admixed population of Atlantic salmon (Salmo salar). Front. Genet. 2014, 5, 402. [Google Scholar]
- Robledo, D.; Matika, O.; Hamilton, A.; Houston, R.D. Genome-Wide Association and Genomic Selection for Resistance to Amoebic Gill Disease in Atlantic Salmon. G3 Genes Genomes Genet. 2018, 8, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-Y.; Hamilton, A.; Tinch, A.E.; Guy, D.R.; Gharbi, K.; Stear, M.; Matika, O.; Bishop, S.C.; Houston, R.D. Genome wide association and genomic prediction for growth traits in juvenile farmed Atlantic salmon using a high density SNP array. BMC Genom. 2015, 16, 969. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, R.L.; Leeds, T.D.; Gao, G.; Parsons, J.E.; Martin, K.; Evenhuis, J.P.; Fragomeni, B.; Wiens, G.D.; Palti, Y. Genomic selection models double the accuracy of predicted breeding values for bacterial cold water disease resistance compared to a traditional pedigree-based model in rainbow trout aquaculture. Genet. Sel. Evol. 2017, 49, 17. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.M.; Bangera, R.; Carvalheiro, R.; Correa, K.; Figueroa, R.; Lhorente, J.P.; Yáñez, J.M. Genomic prediction accuracy for resistance against Piscirickettsia salmonis in farmed rainbow trout. G3 Genes Genomes Genet. 2018, 8, 719–726. [Google Scholar] [CrossRef]
- Palaiokostas, C.; Kocour, M.; Prchal, M.; Houston, R.D. Accuracy of Genomic Evaluations of Juvenile Growth Rate in Common Carp (Cyprinus carpio) Using Genotyping by Sequencing. Front. Genet. 2018, 9, 82. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, Y.; Li, F.; Zhang, X.; Xiang, J. Predictive ability of genomic selection models for breeding value estimation on growth traits of Pacific white shrimp Litopenaeus vannamei. Chin. J. Oceanol. Limnol. 2016, 35, 1221–1229. [Google Scholar] [CrossRef]
- Druet, T.; MacLeod, I.M.; Hayes, B. Toward genomic prediction from whole-genome sequence data: Impact of sequencing design on genotype imputation and accuracy of predictions. Heredity 2013, 112, 39–47. [Google Scholar] [CrossRef]
- Houston, R.D.; Bean, T.P.; MacQueen, D.J.; Gundappa, M.K.; Jin, Y.H.; Jenkins, T.L.; Selly, S.L.C.; Martin, S.A.M.; Stevens, J.R.; Santos, E.M.; et al. Harnessing genomics to fast-track genetic improvement in aquaculture. Nat. Rev. Genet. 2020, 21, 389–409. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Traits | Whole Weight (g) | Shell Length (cm) | Shell Width (cm) | Shell Depth (cm) | Shell Weight (g) | Moisture Content (%) | Taste | Tenderness | Polydora sp. | Marteilioides chungmuensis |
|---|---|---|---|---|---|---|---|---|---|---|
| Whole weight (g) | 0.51 (0.04) | 0.47 (0.04) | 0.51 (0.04) | 0.89 (0.01) | 0.58 (0.04) | n.e | n.e | 0.34 (0.02) | 0.27 (0.03) | |
| Shell length (cm) | 0.67 (0.09) | 0.50 (0.07) | 0.28 (0.05) | 0.16 (0.06) | 0.50 (0.04) | 0.26 (0.05) | 0.06 (0.03) | 0.10 (0.05) | n.e | n.e |
| Shell width (cm) | 0.58 (0.10) | 0.58 (0.11) | 0.55 (0.06) | 0.34 (0.05) | 0.45 (0.04) | 0.26 (0.05) | 0.13 (0.05) | 0.02 (0.01) | n.e | n.e |
| Shell depth (cm) | 0.63 (0.13) | 0.50 (0.10) | 0.84 (0.11) | 0.28 (0.08) | 0.48 (0.04) | 0.16 (0.06) | n.e | n.e | n.e | n.e |
| Shell weight (g) | 0.90 (0.03) | 0.70 (0.08) | 0.54 (0.10) | 0.63 (0.12) | 0.42 (0.07) | 0.04 (0.01) | n.e | n.e | 0.03 (0.01) | n.e |
| Moisture content (%) | 0.60 (0.12) | 0.11 (0.04) | 0.36 (0.12) | 0.07 (0.02) | 0.06 (0.01) | 0.44 (0.07) | 0.12 (0.05) | n.e | n.e | n.e |
| Taste | n.e | 0.07 (0.01) | 0.19 (0.05) | n.e | n.e | 0.14 (0.02) | 0.59 (0.06) | 0.07 (0.02) | 0.02 (0.01) | n.e |
| Tenderness | n.e | 0.12 (0.06) | 0.06 (0.01) | n.e | n.e | 0.07 (0.02) | 0.15 (0.05) | 0.72 (0.06) | n.e | n.e |
| Polydora sp. | 0.37 (0.02) | n.e | n.e | n.e | 0.04 (0.01) | n.e | 0.07 (0.02) | n.e | 0.11 (0.04) | 0.23 (0.03) |
| Marteilioides chungmuensis | 0.35 (0.05) | n.e | n.e | n.e | n.e | n.e | n.e | n.e | 0.66 (0.12) | 0.10 (0.05) |
| Methods | Traits | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Shell Length (cm) | Shell Width (cm) | Shell Depth (cm) | Shell Weight (g) | Moisture Content (%) | Tenderness | Taste | Polydora sp. | Marteilioides chungmuensis | |
| BayesA | 0.734 ± 0.012 | 0.747 ± 0.101 | 0.730 ± 0.014 | 0.780 ± 0.023 | 0.475 ± 0.122 | 0.577 ± 0.025 | 0.485 ± 0.035 | 0.287 ± 0.016 | 0.242 ± 0.016 |
| BayesCπ | 0.732 ± 0.013 | 0.748 ± 0.103 | 0.695 ± 0.012 | 0.766 ± 0.025 | 0.523 ± 0.012 | 0.526 ± 0.021 | 0.481 ± 0.062 | 0.295 ± 0.021 | 0.241 ± 0.021 |
| GBLUP | 0.751 ± 0.024 | 0.750 ± 0.123 | 0.677 ± 0.015 | 0.794 ± 0.124 | 0.504 ± 0.020 | 0.599 ± 0.078 | 0.488 ± 0.065 | 0.300 ± 0.035 | 0.240 ± 0.035 |
| Method | Approach | SNP | Accuracy | Approach | SNP | Accuracy |
|---|---|---|---|---|---|---|
| PBLUP | Pedigree | - | 0.64 | - | - | - |
| GBLUP | MAF 0.01 | 11,492 | 0.76 | Random | 11,492 | 0.76 |
| GBLUP | MAF 0.05 | 9942 | 0.75 | Random | 9942 | 0.75 |
| GBLUP | MAF 0.10 | 7977 | 0.75 | Random | 7977 | 0.75 |
| GBLUP | MAF 0.15 | 6588 | 0.75 | Random | 6588 | 0.74 |
| GBLUP | MAF 0.25 | 4592 | 0.75 | Random | 4592 | 0.74 |
| GBLUP | MAF 0.35 | 3045 | 0.74 | Random | 3045 | 0.73 |
| GBLUP | MAF 0.45 | 1465 | 0.71 | Random | 1465 | 0.70 |
| GBLUP | MAF 0.49 | 440 | 0.67 | Random | 440 | 0.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, S.V.; Gondro, C.; Nguyen, N.T.H.; Gilmour, A.R.; Tearle, R.; Knibb, W.; Dove, M.; Vu, I.V.; Khuong, L.D.; O’Connor, W. Prediction Accuracies of Genomic Selection for Nine Commercially Important Traits in the Portuguese Oyster (Crassostrea angulata) Using DArT-Seq Technology. Genes 2021, 12, 210. https://doi.org/10.3390/genes12020210
Vu SV, Gondro C, Nguyen NTH, Gilmour AR, Tearle R, Knibb W, Dove M, Vu IV, Khuong LD, O’Connor W. Prediction Accuracies of Genomic Selection for Nine Commercially Important Traits in the Portuguese Oyster (Crassostrea angulata) Using DArT-Seq Technology. Genes. 2021; 12(2):210. https://doi.org/10.3390/genes12020210
Chicago/Turabian StyleVu, Sang V., Cedric Gondro, Ngoc T. H. Nguyen, Arthur R. Gilmour, Rick Tearle, Wayne Knibb, Michael Dove, In Van Vu, Le Duy Khuong, and Wayne O’Connor. 2021. "Prediction Accuracies of Genomic Selection for Nine Commercially Important Traits in the Portuguese Oyster (Crassostrea angulata) Using DArT-Seq Technology" Genes 12, no. 2: 210. https://doi.org/10.3390/genes12020210
APA StyleVu, S. V., Gondro, C., Nguyen, N. T. H., Gilmour, A. R., Tearle, R., Knibb, W., Dove, M., Vu, I. V., Khuong, L. D., & O’Connor, W. (2021). Prediction Accuracies of Genomic Selection for Nine Commercially Important Traits in the Portuguese Oyster (Crassostrea angulata) Using DArT-Seq Technology. Genes, 12(2), 210. https://doi.org/10.3390/genes12020210