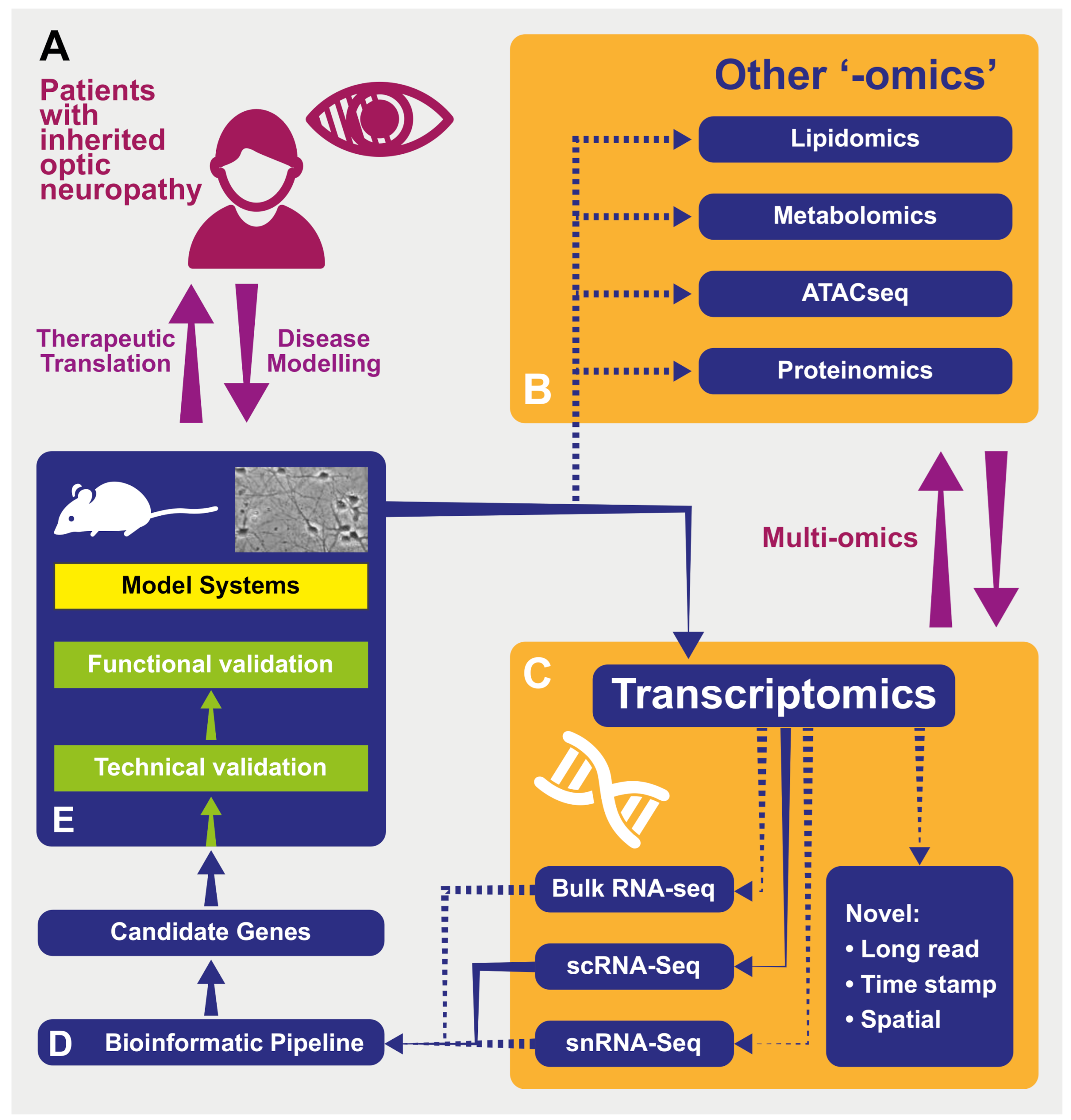
From Transcriptomics to Treatment in Inherited Optic Neuropathies

 ,
,  ,
,  and
and
Abstract
1. Introduction
1.1. Optic Neuropathies
1.2. “-Omics” Technologies as Applied to Inherited Optic Neuropathies
1.3. Transcriptomics
2. Transcriptomics in Inherited Optic Neuropathies
2.1. Applications of Transcriptomics in Optic Neuropathies
2.2. Disadvantages of Transcriptomics in Optic Neuropathies
3. Transcriptomic Methodologies
3.1. Tissue Selection and Sample Preparation
3.2. Quantifying Expression
3.2.1. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
3.2.2. RNA-Seq
3.2.3. Future Directions in Quantifying Expression
3.3. Analysis of RNA-Seq Data
4. Clinical and Research Applications of Transcriptomics
4.1. Diagnostic
4.2. Disease Mechanisms
4.2.1. Exploring Disease Mechanisms Using Transcriptomics
4.2.2. Technical Validation
4.2.3. Functional Validation
4.3. Therapeutic Development
4.3.1. Personalized Medicine
4.3.2. Transferable Neuroprotective Strategies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Carelli, V.; Taiel, M.; Yu-Wai-Man, P. Visual Outcomes in Leber Hereditary Optic Neuropathy Patients with the m.11778G>A (MTND4) Mitochondrial DNA Mutation. J. Neuro-Ophthalmol. 2020, 40, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Brown, D.T.; Howell, N.; Turnbull, D.M.; Chinnery, P.F. The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Burke, A.; Sellar, P.W.; Clarke, M.P.; Gnanaraj, L.; Ah-Kine, D.; Hudson, G.; Czermin, B.; Taylor, R.W.; et al. The Prevalence and Natural History of Dominant Optic Atrophy Due to OPA1 Mutations. Ophthalmology 2010, 117, 1538–1546.e1. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies—Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef]
- Bahr, T.; Welburn, K.; Donnelly, J.; Bai, Y. Emerging model systems and treatment approaches for Leber’s hereditary optic neuropathy: Challenges and opportunities. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165743. [Google Scholar] [CrossRef]
- Coussa, R.G.; Merat, P.; Levin, L.A. Propagation and Selectivity of Axonal Loss in Leber Hereditary Optic Neuropathy. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Gill, K.P.; Hung, S.S.C.; Sharov, A.; Lo, C.Y.; Needham, K.; Lidgerwood, G.E.; Jackson, S.; Crombie, D.E.; Nayagam, B.A.; Cook, A.L.; et al. Enriched retinal ganglion cells derived from human embryonic stem cells. Sci. Rep. 2016, 6, 30552. [Google Scholar] [CrossRef]
- Jankauskaitė, E.; Bartnik, E.; Kodroń, A. Investigating Leber’s hereditary optic neuropathy: Cell models and future perspectives. Mitochondrion 2017, 32, 19–26. [Google Scholar] [CrossRef]
- Rani, L.; Mondal, A.C. Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: Pathogenic and therapeutic implications. Mitochondrion 2020, 50, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Luo, H.; Zhang, K. Leber hereditary optic neuropathy and oxidative stress. Proc. Natl. Acad. Sci. USA 2012, 109, 19882–19883. [Google Scholar] [CrossRef]
- Wallace, D.C.; Lott, M.T. Leber Hereditary Optic Neuropathy: Exemplar of an mtDNA Disease. In Pharmacology of Mitochondria; Singh, H., Sheu, S.-S., Eds.; Springer International: Cham, Switzerland, 2017; pp. 339–376. [Google Scholar] [CrossRef]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 2017, 88, 131–142. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.-M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Ham, M.; Han, J.; Osann, K.; Smith, M.; Kimonis, V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion 2019, 46, 262–269. [Google Scholar] [CrossRef]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Yan, G.; Wang, P.; Wang, X. Mass spectrometry-based metabolomics: Applications to biomarker and metabolic pathway research. Biomed. Chromatogr. 2016, 30, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Esterhuizen, K.; Van Der Westhuizen, F.H.; Louw, R. Metabolomics of mitochondrial disease. Mitochondrion 2017, 35, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kappler, L.; Lehmann, R. Mass-spectrometric multi-omics linked to function—State-of-the-art investigations of mitochondria in systems medicine. TrAC Trends Anal. Chem. 2019, 119, 115635. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A.J. Multi-omics approaches to disease. Genome Biol. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Owen, N.; Moosajee, M. RNA-sequencing in ophthalmology research: Considerations for experimental design and analysis. Ther. Adv. Ophthalmol. 2019, 11, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Masland, R.H. The Neuronal Organization of the Retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Riazifar, H.; Minxin, G.; Huang, T. Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res. Ther. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. OPA1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef]
- Prusky, G.T.; West, P.W.; Douglas, R.M. Behavioral assessment of visual acuity in mice and rats. Vis. Res. 2000, 40, 2201–2209. [Google Scholar] [CrossRef]
- Jagannath, A.; Hughes, S.; Abdelgany, A.; Pothecary, C.A.; Di Pretoro, S.; Pires, S.S.; Vachtsevanos, A.; Pilorz, V.; Brown, L.A.; Hossbach, M.; et al. Isoforms of Melanopsin Mediate Different Behavioral Responses to Light. Curr. Biol. 2015, 25, 2430–2434. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.R.; Issa, P.C.; Perganta, G.; Williams, P.A.; Davies, V.J.; Sekaran, S.; Votruba, M.; MacLaren, R.E. Specific deficits in visual electrophysiology in a mouse model of dominant optic atrophy. Exp. Eye Res. 2011, 93, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-M.; Shafi, A.; Nguyen, T.; Draghici, S. Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Gilhooley, M.J.; Hickey, D.G.; Hughes, S.; Hankins, M.W. Retinal bipolar cell gene changes in the rd1 model of inherited retinal degeneration. In Proceedings of ARVO Annual Meeting Abstract, Honolulu, HI, USA, 29 April 2018; Investigative Ophthalmology & Visual Science: Washington, DC, USA, 2018; Volume 59, 1007p. [Google Scholar]
- Lozano, D.C.; Choi, D.; Jayaram, H.; Morrison, J.C.; Johnson, E.C. Utilizing RNA-Seq to Identify Differentially Expressed Genes in Glaucoma Model Tissues, Such as the Rodent Optic Nerve Head. In Advanced Structural Safety Studies; Springer: Singapore, 2017; Volume 1695, pp. 299–310. [Google Scholar]
- Yu-Wai-Man, C.; Owen, N.; Lees, J.; Tagalakis, A.D.; Hart, S.L.; Webster, A.R.; Orengo, C.A.; Khaw, P.T. Genome-wide RNA-Sequencing analysis identifies a distinct fibrosis gene signature in the conjunctiva after glaucoma surgery. Sci. Rep. 2017, 7, 5644. [Google Scholar] [CrossRef] [PubMed]
- Parekh, S.; Ziegenhain, C.; Vieth, B.; Enard, W.; Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 2016, 6, 25533. [Google Scholar] [CrossRef]
- Zhao, W.; He, X.; Hoadley, K.A.; Parker, J.S.; Hayes, D.N.; Perou, C.M. Comparison of RNA-Seq by poly(A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genom. 2014, 15, 1–11. [Google Scholar] [CrossRef]
- O’Neil, D.; Glowatz, H.; Schlumpberger, M. Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. Curr. Protoc. Mol. Biol. 2013, 103, 1–8. [Google Scholar] [CrossRef]
- Peirson, S.N. Quantitative analysis of ocular gene expression. In Real-Time PCR, 1st ed.; Taylor & Francis: Abingdon, UK, 2007; pp. 135–154. [Google Scholar]
- Wang, W.; Mcnatt, L.G.; Shepard, A.; Jacobson, N.; Nishimura, D.; Stone, E.; Sheffield, V.; Clark, A. Optimal procedure for extracting RNA from human ocular tissues and expression profiling of the congenital glaucoma gene FOXC1 using quantitative RT-PCR. Mol. Vis. 2001, 7, 89–94. [Google Scholar]
- Peirson, S.N.; Butler, J.N.; Foster, R.G. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003, 31, e73. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative Monitoring of Gene Expression Patterns with a Complementary DNA Microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J. The beginning of the end for microarrays? Nat. Methods 2008, 5, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Bumgarner, R. Overview of DNA Microarrays: Types, Applications, and Their Future. In Current Protocols in Molecular Biology; Wiley-Blackwell: Hoboken, NJ, USA, 2013; Volume 101. [Google Scholar] [CrossRef]
- Cieślik, M.; Chinnaiyan, A.M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2018, 19, 93–109. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Martersteck, E.M.; Hirokawa, K.E.; Evarts, M.; Bernard, A.; Duan, X.; Li, Y.; Ng, L.; Oh, S.W.; Ouellette, B.; Royall, J.J.; et al. Diverse Central Projection Patterns of Retinal Ganglion Cells. Cell Rep. 2017, 18, 2058–2072. [Google Scholar] [CrossRef]
- Baden, T.; Berens, P.; Franke, K.J.; Rosón, M.R.; Bethge, M.; Euler, T. The functional diversity of retinal ganglion cells in the mouse. Nature 2016, 529, 345–350. [Google Scholar] [CrossRef]
- Rheaume, B.A.; Jereen, A.; Bolisetty, M.; Sajid, M.S.; Yang, Y.; Renna, K.; Sun, L.; Robson, P.; Trakhtenberg, E.F. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun. 2018, 9, 2759. [Google Scholar] [CrossRef]
- Cui, Q.; Ren, C.; Sollars, P.J.; Pickard, G.E.; So, K.-F. The injury resistant ability of melanopsin-expressing intrinsically photosensitive retinal ganglion cells. Neuroscience 2015, 284, 845–853. [Google Scholar] [CrossRef]
- Kulkarni, A.; Anderson, A.G.; Merullo, D.P.; Konopka, G. Beyond bulk: A review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 2019, 58, 129–136. [Google Scholar] [CrossRef]
- Aldridge, S.; Teichmann, S.A. Single cell transcriptomics comes of age. Nat. Commun. 2020, 11, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Codeluppi, S.; Yung, Y.C.; Gao, D.; Chun, J.; Kharchenko, P.; Linnarsson, S.; Zhang, K. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kashima, Y.; Sakamoto, Y.; Kaneko, K.; Seki, M.; Suzuki, Y.; Suzuki, A. Single-cell sequencing techniques from individual to multiomics analyses. Exp. Mol. Med. 2020, 52, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.M.; Shekhar, K.; Whitney, I.E.; Jacobi, A.; Benhar, I.; Hong, G.; Yan, W.; Adiconis, X.; Arnold, M.E.; Lee, J.M.; et al. Single-Cell Profiles of Retinal Ganglion Cells Differing in Resilience to Injury Reveal Neuroprotective Genes. Neuron 2019, 104, 1039–1055.e12. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Chen, L.M.; Liu, S.; Zhong, E.D.; Scherrer, J.R.; Boyden, E.S.; Chen, F. RNA timestamps identify the age of single molecules in RNA sequencing. Nat. Biotechnol. 2020, 1–6. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-cell multiomics: Technologies and data analysis methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, E.N. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217.e12. [Google Scholar] [CrossRef]
- Reinhard, K.; Tikidji-Hamburyan, A.; Seitter, H.; Idrees, S.; Mutter, M.; Benkner, B.; Münch, T.A. Step-By-Step Instructions for Retina Recordings with Perforated Multi Electrode Arrays. PLoS ONE 2014, 9, e106148. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 1–19. [Google Scholar] [CrossRef]
- Vieth, B.; Parekh, S.; Ziegenhain, C.; Enard, W.; Hellmann, I. A systematic evaluation of single cell RNA-seq analysis pipelines. Nat. Commun. 2019, 10, 4667. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed]
- McDermaid, A.; Monier, B.; Zhao, J.; Liu, B.; Ma, Q. Interpretation of differential gene expression results of RNA-seq data: Review and integration. Brief. Bioinform. 2019, 20, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.J. Precision Medicine in Practice: Molecular Diagnosis Enabling Precision Therapies. Clin. Lab. Med. 2020, 40, 113–230. [Google Scholar] [CrossRef]
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Oláhová, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2020, 43, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Prokisch, H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018, 62, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Frésard, L.; Smail, C.; Ferraro, N.M.; Teran, N.A.; Li, X.; Smith, K.S.; Bonner, D.; Kernohan, K.D.; Marwaha, S.; Zappala, Z.; et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med. 2019, 25, 911–919. [Google Scholar] [CrossRef]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 1–10. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14–R12. [Google Scholar] [CrossRef] [PubMed]
- Rahmatallah, Y.; Emmert-Streib, F.; Glazko, G. Gene set analysis approaches for RNA-seq data: Performance evaluation and application guideline. Brief. Bioinform. 2015, 17, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Rotroff, D.; Ma, J.; Shojaie, A.; Motsinger-Reif, A. Gene set analysis methods: A systematic comparison. BioData Min. 2018, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Elstner, M.; Turnbull, D.M. Transcriptome analysis in mitochondrial disorders. Brain Res. Bull. 2012, 88, 285–293. [Google Scholar] [CrossRef]
- Danielson, S.R.; Carelli, V.; Tan, G.; Martinuzzi, A.; Schapira, A.H.V.; Savontaus, M.-L.; Cortopassi, G. Isolation of transcriptomal changes attributable to LHON mutations and the cybridization process. Brain 2005, 128, 1026–1037. [Google Scholar] [CrossRef]
- Cortopassi, G.; Danielson, S.; Alemi, M.; Zhan, S.S.; Tong, W.; Carelli, V.; Martinuzzi, A.; Marzuki, S.; Majamaa, K.; Wong, A. Mitochondrial disease activates transcripts of the unfolded protein response and cell cycle and inhibits vesicular secretion and oligodendrocyte-specific transcripts. Mitochondrion 2006, 6, 161–175. [Google Scholar] [CrossRef]
- Yu, A.K.; Song, L.; Murray, K.D.; Van Der List, D.; Sun, C.; Shen, Y.; Xia, Z.; Cortopassi, G. Mitochondrial complex I deficiency leads to inflammation and retinal ganglion cell death in the Ndufs4 mouse. Hum. Mol. Genet. 2015, 24, 2848–2860. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, M.; Kaushik, A.M.; Chang, X.; Duan, Y.; Chen, L.; Wang, J.; Berlinicke, C.; Zack, D.J. Single-Cell Transcriptome Profiling of Human Stem Cell-Derived Retinal Ganglion Cells in a Dominant Optic Atrophy Model. In Proceedings of ARVO Annual Meeting Abstract, Honolulu, HI, USA, 29 April 2018; Investigative Ophthalmology & Visual Science: Washington, DC, USA, 2018; Volume 59, 1998p. [Google Scholar]
- Wu, Y.-R.; Wang, A.-G.; Chen, Y.-T.; Yarmishyn, A.A.; Buddhakosai, W.; Yang, T.-C.; Hwang, D.-K.; Yang, Y.-P.; Shen, C.-N.; Lee, H.-C.; et al. Bioactivity and gene expression profiles of hiPSC-generated retinal ganglion cells in MT-ND4 mutated Leber’s hereditary optic neuropathy. Exp. Cell Res. 2018, 363, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, S.; Hashim, A.; Cieslar-Pobuda, A.; Jensen, V.; Behringer, S.; Talug, B.; Chu, D.T.; Pecquet, C.; Rogne, M.; Brech, A.; et al. Optic Atrophy 1 Controls Human Neuronal Development by Preventing Aberrant Nuclear DNA Methylation. iScience 2020, 23, 101154. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sant, D.W.; Wang, G.; Guy, J. Mitochondrial Transfer of the Mutant Human ND6T14484C Gene Causes Visual Loss and Optic Neuropathy. Transl. Vis. Sci. Technol. 2020, 9, 1. [Google Scholar] [CrossRef]
- Fang, Z.; Cui, X. Design and validation issues in RNA-seq experiments. Brief. Bioinform. 2011, 12, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Nair, K.S. Mitochondrial metabolic function assessed in vivo and in vitro. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.R.; Bennett, J.; Wellman, J.; Do, D.C.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Douglas, R.M.; Alam, N.M.; Silver, B.D.; McGill, T.J.; Tschetter, W.W.; Prusky, G.T. Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis. Neurosci. 2005, 22, 677–684. [Google Scholar] [CrossRef]
- Safety Evaluation of Gene Therapy in Leber Hereditary Optic Neuropathy (LHON) Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02064569 (accessed on 22 January 2021).
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.C.; Rudolph, G.; et al. Bilateral visual improvement with unilateral gene therapy for Leber hereditary optic neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Ratican, S.E.; Osborne, A.; Martin, K.R. Progress in Gene Therapy to Prevent Retinal Ganglion Cell Loss in Glaucoma and Leber’s Hereditary Optic Neuropathy. Neural Plast. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Osborne, A.; Wang, A.X.; Tassoni, A.; Widdowson, P.S.; Martin, K.R. Design of a Novel Gene Therapy Construct to Achieve Sustained Brain-Derived Neurotrophic Factor Signaling in Neurons. Hum. Gene Ther. 2018, 29, 828–841. [Google Scholar] [CrossRef] [PubMed]
- de Silva, S.R.; McClements, M.E.; Hankins, M.W.; MacLaren, R.E. Adeno-Associated Viral Gene Therapy for Retinal Disorders. In Gene Delivery and Therapy for Neurological Disorders; Bo, X., Verhaagen, J., Eds.; Springer: New York, NY, USA, 2015; Volume 98, pp. 203–228. [Google Scholar]
- Moster, M.; Sadun, A.; Klopstock, T.; Newman, N.; Vignal-Clermont, C.; Carelli, V.; Yu-Wai-Man, P.; Biousse, V.; Sergott, R.; Katz, B.; et al. rAAV2/2-ND4 for the Treatment of Leber Hereditary Optic Neuropathy (LHON): Final Results from the RESCUE and REVERSE Phase III Clinical Trials and Experimental Data in Nonhuman Primates to Support a Bilateral Effect (2339). Neurology 2020, 94, 2339. [Google Scholar]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Biousse, V.; Sadun, A.A.; Moster, M.L.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Blouin, L.; et al. Bilateral Visual Improvement with Unilateral Gene Therapy for Leber Hereditary Optic Neuropathy (LHON). Invest. Ophthalmol. Vis. Sci. 2020, 61, 5181. [Google Scholar]
- Rossmiller, B.; Mao, H.; Lewin, A.S. Gene therapy in animal models of autosomal dominant retinitis pigmentosa. Mol. Vis. 2012, 18, 2479–2496. [Google Scholar] [PubMed]
- Orlans, H.O.; McClements, M.E.; Barnard, A.R.; Martinez-Fernandez, d.C.; MacLaren, R.E. Mirtron gene therapy for the treatment of rhodopsin-related dominant retinitis pigmentosa. In Proceedings of ARVO Annual Meeting Abstract, Vancouver, BC, Canada, 30 April 2019; Investigative Ophthalmology & Visual Science: Wahington, DC, USA, 2019; Volume 60. [Google Scholar]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef]
- Massengill, M.T.; Young, B.M.; Lewin, A.S.; Ildefonso, C.J. Co-Delivery of a Short-Hairpin RNA and a shRNA-Resistant Replacement Gene with Adeno-Associated Virus: An Allele-Independent Strategy for Autosomal-Dominant Retinal Disorders. In Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 235–258. [Google Scholar] [CrossRef]
- Tribble, J.R.; Otmani, A.; Sun, S.; Ellis, S.A.; Cimaglia, G.; Vohra, R.; Joe, M.; Lardner, E.; Venkataraman, A.P.; Dominguez-Vicent, A.; et al. Nicotinamide provides neuroprotection in glaucoma by protecting against mitochondrial and metabolic dysfunction. bioRxiv 2020, 2020.10.21.348250. Available online: https://www.biorxiv.org/content/10.1101/2020.10.21.348250v1 (accessed on 22 January 2021).
- Fu, L.; Kwok, S.S.; Chan, Y.-K.; Lai, J.S.M.; Pan, W.; Nie, L.; Shih, K.C. Therapeutic Strategies for Attenuation of Retinal Ganglion Cell Injury in Optic Neuropathies: Concepts in Translational Research and Therapeutic Implications. BioMed Res. Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Martin, K.; Martin, K.R. Neuroprotection in Glaucoma: Towards Clinical Trials and Precision Medicine. Curr. Eye Res. 2020, 45, 327–338. [Google Scholar] [CrossRef]
- Pasi, K.J.; Rangarajan, S.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Laffan, M.; Russell, C.B.; Li, M.; Pierce, G.F.; et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N. Engl. J. Med. 2020, 382, 29–40. [Google Scholar] [CrossRef]


{kind=link}
| Reference | Condition | Model Used | Control | Technique | Conclusion |
|---|---|---|---|---|---|
| Danielson 2005 [83] | LHON | Cybrid cell lines containing mt11778 mutation | Cybrid cell lines without mt11778 mutation | Affymetrix U95Av2 oligonucleotide microarray | 96 differently expressed genes, but only 9 of these replicated in other models. |
| Cortopassi 2006 [84] | LHON | Multiple cell lines carrying mt3460, 11778 or 14484 mutation for LHON as well as other cell lines corresponding to other common mitochondrial diseases | Corresponding cell lines without LHON causing mutations. | Affymetrix U95Av2 oligonucleotide microarray | Across models of multiple mitochondrial diseases, unfolded protein response and cell cycle pathways are upregulated and those involving vesicular secretion and protein synthesis are downregulated. |
| Yu 2015 [85] | LHON/complex I deficiency | Ndufs4 knock out mouse | Wild-type mice | Whole retina bulk RNA-seq | At whole retina level, genes most dramatically over represented are those associated with innate immunity and inflammation. |
| Cheng 2018 [86] | OPA1-DOA | Cultured RGCs derived from human stem cells with mutations in OPA1 introduced by CRISPR | Cultured RGCs derived from human stem cells without mutations | scRNA-seq | Exploratory study. Pathways involved in RGC fate specification, axon guidance, and regeneration upregulated in OPA1-mutated cells. |
| Wu 2018 [87] | LHON | Cultured RGCs derived from iPSC from a LHON patient (mt11778 mutation) | Cultured RGCs derived from an asymptomatic carrier and control | GeneChip Human Genome U133 Plus 2.0 oligonucleotide microarrays | Genes implicated in “cell cycle” and extracellular matrix most over represented. |
| Calayan 2020 [88] | OPA1-DOA | Cultured neurons heterozygous for OPA1 KO Cultured patient induced neural progenitor cells (c.2873_2876delTTAG) | Corresponding cells without mutation | Bulk RNA-seq | Downregulation of genes important for GABAergic neurons and retinal development. |
| Yu 2020 [89] | LHON | Optic nerve tissue from DBA1/J mice intravitreally injected with AAV coding for human ND6 T14484C mutation | Optic nerve tissue from uninfected eyes | Bulk RNA-seq | Marked changes in gene expression, with pathways relating to oxidative stress and apoptosis particularly represented. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilhooley, M.J.; Owen, N.; Moosajee, M.; Yu Wai Man, P. From Transcriptomics to Treatment in Inherited Optic Neuropathies. Genes 2021, 12, 147. https://doi.org/10.3390/genes12020147
Gilhooley MJ, Owen N, Moosajee M, Yu Wai Man P. From Transcriptomics to Treatment in Inherited Optic Neuropathies. Genes. 2021; 12(2):147. https://doi.org/10.3390/genes12020147
Chicago/Turabian StyleGilhooley, Michael James, Nicholas Owen, Mariya Moosajee, and Patrick Yu Wai Man. 2021. "From Transcriptomics to Treatment in Inherited Optic Neuropathies" Genes 12, no. 2: 147. https://doi.org/10.3390/genes12020147
APA StyleGilhooley, M. J., Owen, N., Moosajee, M., & Yu Wai Man, P. (2021). From Transcriptomics to Treatment in Inherited Optic Neuropathies. Genes, 12(2), 147. https://doi.org/10.3390/genes12020147