
Detection of 46, XY Disorder of Sex Development (DSD) Based on Plasma Cell-Free DNA and Targeted Next-Generation Sequencing

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Report
2.2. DNA Extraction, cffDNA Isolation from Plasma and NIPT Analysis
2.3. DNA Extraction, PCR and Karyotype Analysis
2.4. Target Region Capture Sequencing and Bioinformatics Analysis
2.5. Mutation Validation
3. Results
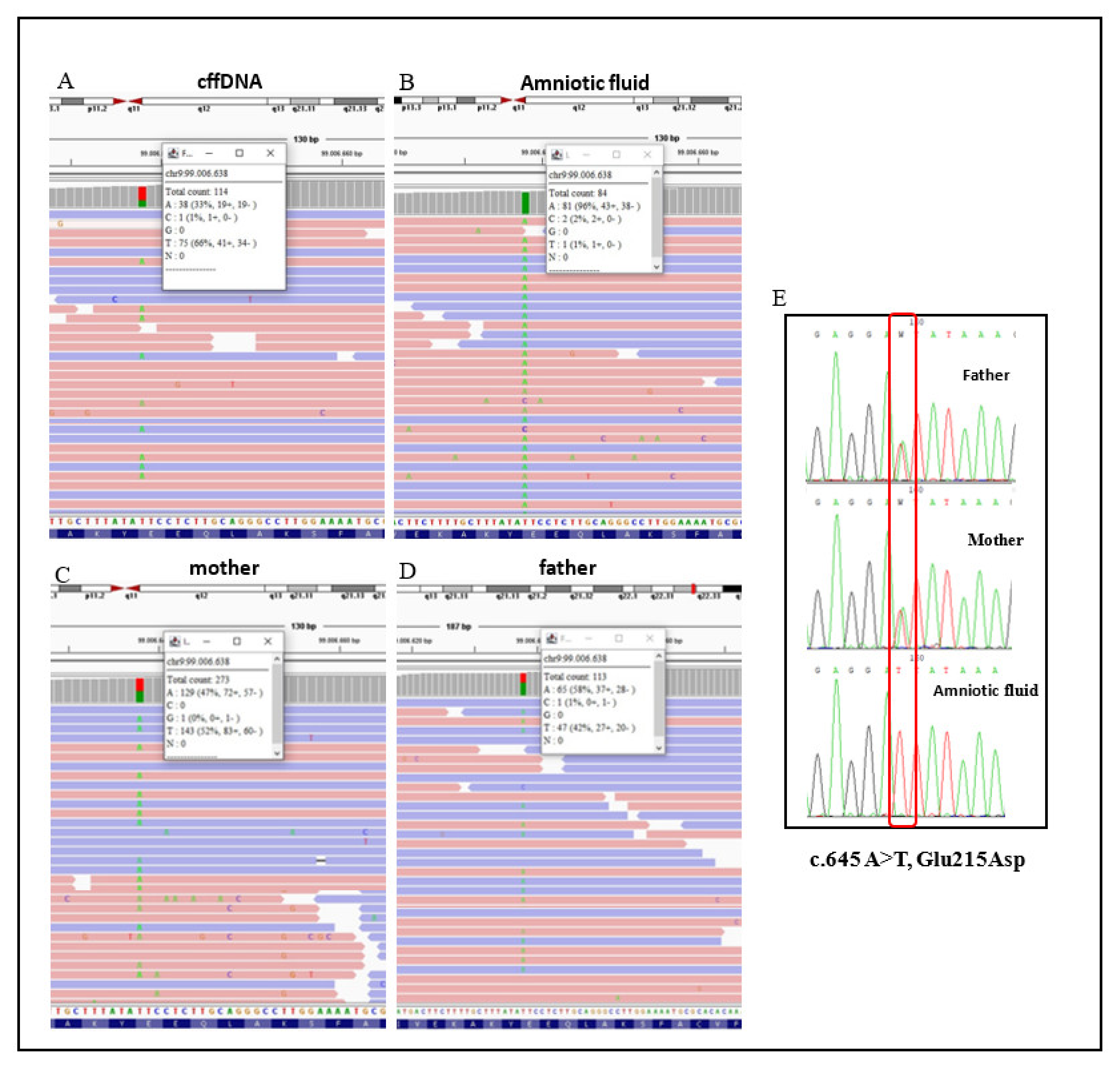
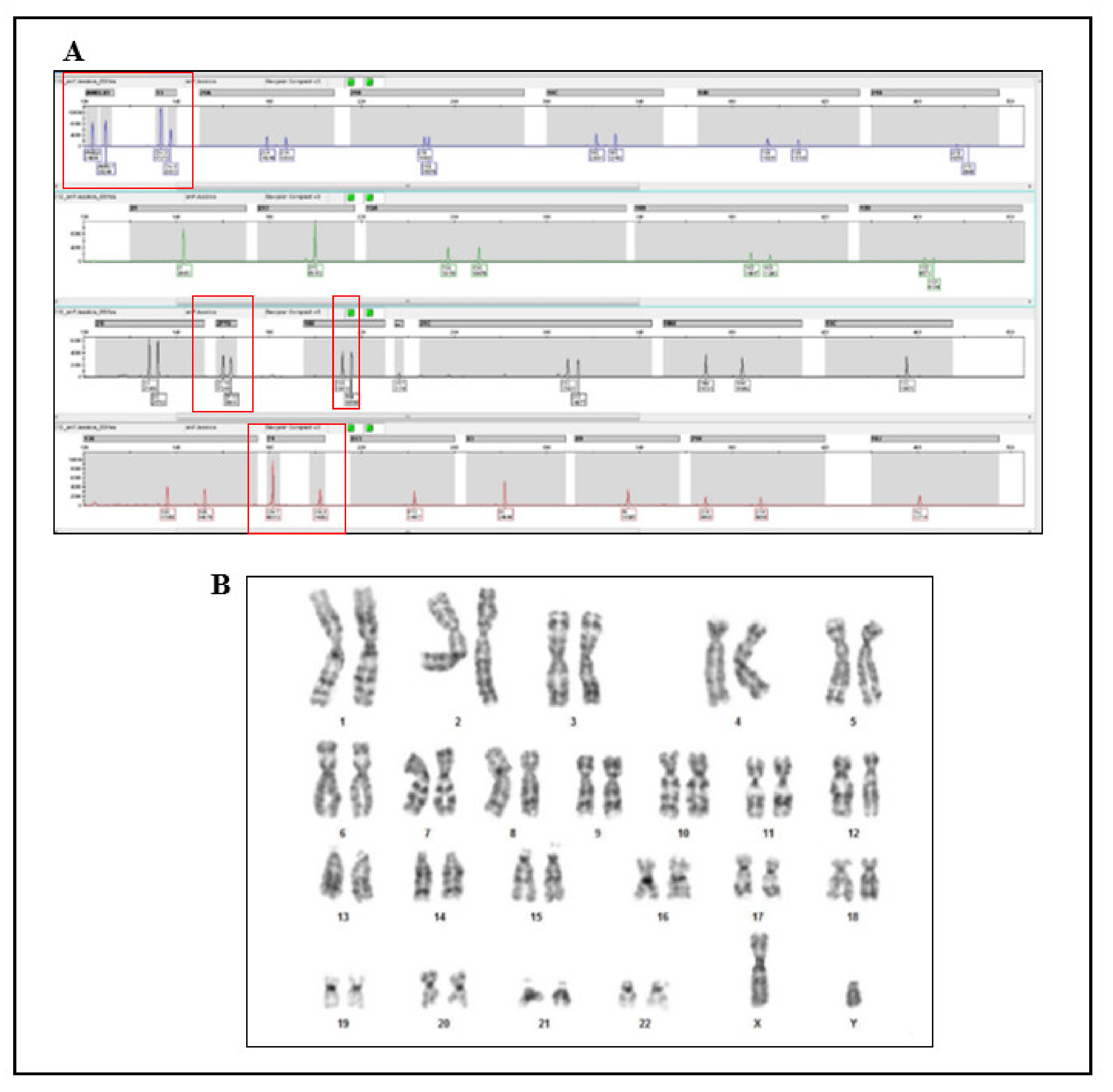
3.1. NIPT Analysis
3.2. Cytogenetic Analysis
3.3. Molecular Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosler, A. 17β-Hydroxysteroid Dehydrogenase 3 Deficiency in the Mediterranean Population. Pediatr. Endocrinol. Rev. 2006, 3 (Suppl. 3), 455–461. [Google Scholar]
- Faienza, M.F.; Giordani, L.; Delvecchio, M.; Cavallo, L. Clinical, Endocrine, and Molecular Findings in 17β -Hydroxysteroid Dehydrogenase Type 3 Deficiency. J. Endocrinol. Investig. 2008, 31, 85–91. [Google Scholar] [CrossRef]
- George, M.M.; New, M.I.; Ten, S.; Sultan, C.; Bhangoo, A. The Clinical and Molecular Heterogeneity of 17βHSD-3 Enzyme Deficiency. Horm. Res. Paediatr. 2010, 74, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Bertelloni, S.; Balsamo, A.; Giordani, L.; Fischetto, R.; Russo, G.; Delvecchio, M.; Gennari, M.; Nicoletti, A.; Maggio, M.C.; Concolino, D.; et al. 17β-Hydroxysteroid Dehydrogenase-3 Deficiency: From Pregnancy to Adolescence. J. Endocrinol. Investig. 2009, 32, 666–670. [Google Scholar] [CrossRef]
- Levy-Khademi, F.; Zeligson, S.; Lavi, E.; Klopstock, T.; Chertin, B.; Avnon-Ziv, C.; Abulibdeh, A.; Renbaum, P.; Rosen, T.; Perlberg-Bengio, S.; et al. The Novel Founder Homozygous V225M Mutation in the HSD17B3 Gene Causes Aberrant Splicing and XY-DSD. Endocrine 2020, 69, 650–654. [Google Scholar] [CrossRef] [PubMed]
- De Castro, C.C.T.S.; Guaragna-Filho, G.; Calais, F.L.; Coeli, F.B.; Leal, I.R.L.; Cavalcante-Junior, E.F.; Monlleó, I.L.; Pereira, S.R.F.; de Silva, R.B.P.; Gabiatti, J.R.E.; et al. Clinical and Molecular Spectrum of Patients with 17β-Hydroxysteroid Dehydrogenase Type 3 (17-β-HSD3) Deficiency. Arq. Bras. Endocrinol. Metab. 2012, 56, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, D.W.; Platt, L.D.; Goldberg, J.D.; Abuhamad, A.Z.; Sehnert, A.J.; Rava, R.P. Genome-Wide Fetal Aneuploidy Detection by Maternal Plasma DNA Sequencing. Obstet. Gynecol. 2012, 119, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.M.; Accurti, V.; Santacruz, B.; Plana, M.N.; Nicolaides, K.H. Analysis of Cell-Free DNA in Maternal Blood in Screening for Aneuploidies: Updated Meta-Analysis: Cell-Free DNA in Screening for Aneuploidies. Ultrasound Obstet. Gynecol. 2017, 50, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Phillips, S.; Freeman, K.; Geppert, J.; Agbebiyi, A.; Uthman, O.A.; Madan, J.; Clarke, A.; Quenby, S.; Clarke, A. Accuracy of Non-Invasive Prenatal Testing Using Cell-Free DNA for Detection of Down, Edwards and Patau Syndromes: A Systematic Review and Meta-Analysis. BMJ Open 2016, 6, e010002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitty, L.; Hui, L.; Ghidini, A.; Levy, B.; Deprest, J.; Van Mieghem, T.; Bianchi, D. In Case You Missed It: The Prenatal Diagnosis Editors Bring You the Most Significant Advances of 2019. Prenat. Diagn. 2020, 40, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitty, L.S.; Mason, S.; Barrett, A.N.; McKay, F.; Lench, N.; Daley, R.; Jenkins, L.A. Non-invasive Prenatal Diagnosis of Achondroplasia and Thanatophoric Dysplasia: Next-generation Sequencing Allows for a Safer, More Accurate, and Comprehensive Approach. Prenat. Diagn. 2015, 35, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, J.; Saucier, J.B.; Feng, Y.; Jiang, Y.; Sinson, J.; McCombs, A.K.; Schmitt, E.S.; Peacock, S.; Chen, S.; et al. Non-Invasive Prenatal Sequencing for Multiple Mendelian Monogenic Disorders Using Circulating Cell-Free Fetal DNA. Nat. Med. 2019, 25, 439–447. [Google Scholar] [CrossRef]
- Hill, M.; Twiss, P.; Verhoef, T.I.; Drury, S.; McKay, F.; Mason, S.; Jenkins, L.; Morris, S.; Chitty, L.S. Non-Invasive Prenatal Diagnosis for Cystic Fibrosis: Detection of Paternal Mutations, Exploration of Patient Preferences and Cost Analysis. Prenat. Diagn. 2015, 35, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.N.; McDonnell, T.C.R.; Chan, K.C.A.; Chitty, L.S. Digital PCR Analysis of Maternal Plasma for Noninvasive Detection of Sickle Cell Anemia. Clin. Chem. 2012, 58, 1026–1032. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.; Barrett, A.N.; Hua, R.; Tan, T.Z.; Ho, S.S.Y.; Chan, J.K.Y.; Zhong, M.; Choolani, M. Non-Invasive Prenatal Diagnostic Testing for β-Thalassaemia Using Cell-Free Fetal DNA and next Generation Sequencing. Prenat. Diagn. 2015, 35, 258–265. [Google Scholar] [CrossRef] [PubMed]
- La Verde, M.; De Falco, L.; Torella, A.; Savarese, G.; Savarese, P.; Ruggiero, R.; Conte, A.; Fico, V.; Torella, M.; Fico, A. Performance of Cell-Free DNA Sequencing-Based Non-Invasive Prenatal Testing: Experience on 36,456 Singleton and Multiple Pregnancies. BMC Med. Genom. 2021, 14, 93. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.; Fox, S.P.; Abbs, S.J.; Yau, S.C.; Scriven, P.N.; Docherty, Z.; Ogilvie, C.M. Development and Implementation of a New Rapid Aneuploidy Diagnostic Service within the UK National Health Service and Implications for the Future of Prenatal Diagnosis. Lancet 2001, 358, 1057–1061. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Hughes, I.A. Consensus Statement on Management of Intersex Disorders. Arch. Dis. Child. 2005, 91, 554–563. [Google Scholar] [CrossRef]
- Bertelloni, S.; Dati, E.; Hiort, O. Diagnosis of 17β-Hydroxysteroid Dehydrogenase Deficiency. Expert Rev. Endocrinol. Metab. 2009, 4, 53–65. [Google Scholar] [CrossRef]
- Boehmer, A.L.M.; Brinkmann, A.O.; Sandkuijl, L.A.; Halley, D.J.J.; Niermeijer, M.F.; Andersson, S.; de Jong, F.H.; Kayserili, H.; de Vroede, M.A.; Otten, B.J.; et al. 17β-Hydroxysteroid Dehydrogenase-3 Deficiency: Diagnosis, Phenotypic Variability, Population Genetics, and Worldwide Distribution of Ancient and de Novo Mutations. J. Clin. Endocrinol. Metab. 1999, 84, 4713–4721. [Google Scholar] [CrossRef] [PubMed]
- Faienza, M.F.; Baldinotti, F.; Marrocco, G.; TyuTyusheva, N.; Peroni, D.; Baroncelli, G.I.; Bertelloni, S. 17β-Hydroxysteroid Dehydrogenase Type 3 Deficiency: Female Sex Assignment and Follow-Up. J. Endocrinol. Investig. 2020, 43, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.; Geissler, W.M.; Wu, L.; Davis, D.L.; Grumbach, M.M.; New, M.I.; Schwarz, H.P.; Blethen, S.L.; Mendonca, B.B.; Bloise, W.; et al. Molecular Genetics and Pathophysiology of 17 Beta-Hydroxysteroid Dehydrogenase 3 Deficiency. J. Clin. Endocrinol. Metab. 1996, 81, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kirk, J.M.W.; Stanhope, R.G.; Johnston, D.I.; Harland, S.; Auchus, R.J.; Andersson, S.; Hughes, I.A. Phenotypic Variability in 17?-Hydroxysteroid Dehydrogenase-3 Deficiency and Diagnostic Pitfalls. Clin. Endocrinol. 2007, 67, 20–28. [Google Scholar] [CrossRef]
- Mendonca, B.B.; Inacio, M.; Arnhold, I.J.P.; Costa, E.M.F.; Bloise, W.; Martin, R.M.; Denes, F.T.; Silva, F.A.Q.; Andersson, S.; Lindqvist, A.; et al. Male Pseudohermaphroditism Due to 17β-Hydroxysteroid Dehydrogenase 3 Deficiency: Diagnosis, Psychological Evaluation, and Management. Medicine 2000, 79, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Khattab, A.; Yuen, T.; Yau, M.; Domenice, S.; Frade Costa, E.M.; Diya, K.; Muhuri, D.; Pina, C.E.; Nishi, M.Y.; Yang, A.C.; et al. Pitfalls in Hormonal Diagnosis of 17-Beta Hydroxysteroid Dehydrogenase III Deficiency. J. Pediatric Endocrinol. Metab. 2015, 28, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.; Chitty, L.S. Beyond Screening for Chromosomal Abnormalities: Advances in Non-Invasive Diagnosis of Single Gene Disorders and Fetal Exome Sequencing. Semin. Fetal Neonatal Med. 2018, 23, 94–101. [Google Scholar] [CrossRef]
- Alberry, M.S.; Aziz, E.; Ahmed, S.R.; Abdel-fattah, S. Non Invasive Prenatal Testing (NIPT) for Common Aneuploidies and Beyond. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 258, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.; Cariati, F.; Sarno, L.; Conforti, A.; Bagnulo, F.; Strina, I.; Pastore, L.; Maruotti, G.M.; Alviggi, C. Non-Invasive Prenatal Testing: Current Perspectives and Future Challenges. Genes 2020, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.M.; Arboleda, V.A.; Lee, H.; Barseghyan, H.; Adam, M.P.; Fechner, P.Y.; Bargman, R.; Keegan, C.; Travers, S.; Schelley, S.; et al. Exome Sequencing for the Diagnosis of 46,XY Disorders of Sex Development. J. Clin. Endocrinol. Metab. 2015, 100, E333–E344. [Google Scholar] [CrossRef]
- Eggers, S.; Sadedin, S.; van den Bergen, J.A.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.M.; Tan, T.Y.; et al. Disorders of Sex Development: Insights from Targeted Gene Sequencing of a Large International Patient Cohort. Genome Biol. 2016, 17, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Falco, L.; Piscopo, C.; D’Angelo, R.; Evangelista, E.; Suero, T.; Sirica, R.; Ruggiero, R.; Savarese, G.; Di Carlo, A.; Furino, G.; et al. Detection of 46, XY Disorder of Sex Development (DSD) Based on Plasma Cell-Free DNA and Targeted Next-Generation Sequencing. Genes 2021, 12, 1890. https://doi.org/10.3390/genes12121890
De Falco L, Piscopo C, D’Angelo R, Evangelista E, Suero T, Sirica R, Ruggiero R, Savarese G, Di Carlo A, Furino G, et al. Detection of 46, XY Disorder of Sex Development (DSD) Based on Plasma Cell-Free DNA and Targeted Next-Generation Sequencing. Genes. 2021; 12(12):1890. https://doi.org/10.3390/genes12121890
Chicago/Turabian StyleDe Falco, Luigia, Carmelo Piscopo, Rossana D’Angelo, Eloisa Evangelista, Teresa Suero, Roberto Sirica, Raffaella Ruggiero, Giovanni Savarese, Antonella Di Carlo, Giulia Furino, and et al. 2021. "Detection of 46, XY Disorder of Sex Development (DSD) Based on Plasma Cell-Free DNA and Targeted Next-Generation Sequencing" Genes 12, no. 12: 1890. https://doi.org/10.3390/genes12121890
APA StyleDe Falco, L., Piscopo, C., D’Angelo, R., Evangelista, E., Suero, T., Sirica, R., Ruggiero, R., Savarese, G., Di Carlo, A., Furino, G., Scarpato, C., & Fico, A. (2021). Detection of 46, XY Disorder of Sex Development (DSD) Based on Plasma Cell-Free DNA and Targeted Next-Generation Sequencing. Genes, 12(12), 1890. https://doi.org/10.3390/genes12121890