
Gene Therapeutic Approaches for the Treatment of Mitochondrial Dysfunction in Parkinson’s Disease


Abstract
1. Introduction
1.1. Mitochondrial Dysfunction in Idiopathic and Monogenic Parkinson’s Disease
1.2. The Scope of This Review
1.3. Current Gene Therapeutic Approaches in Parkinson’s Disease
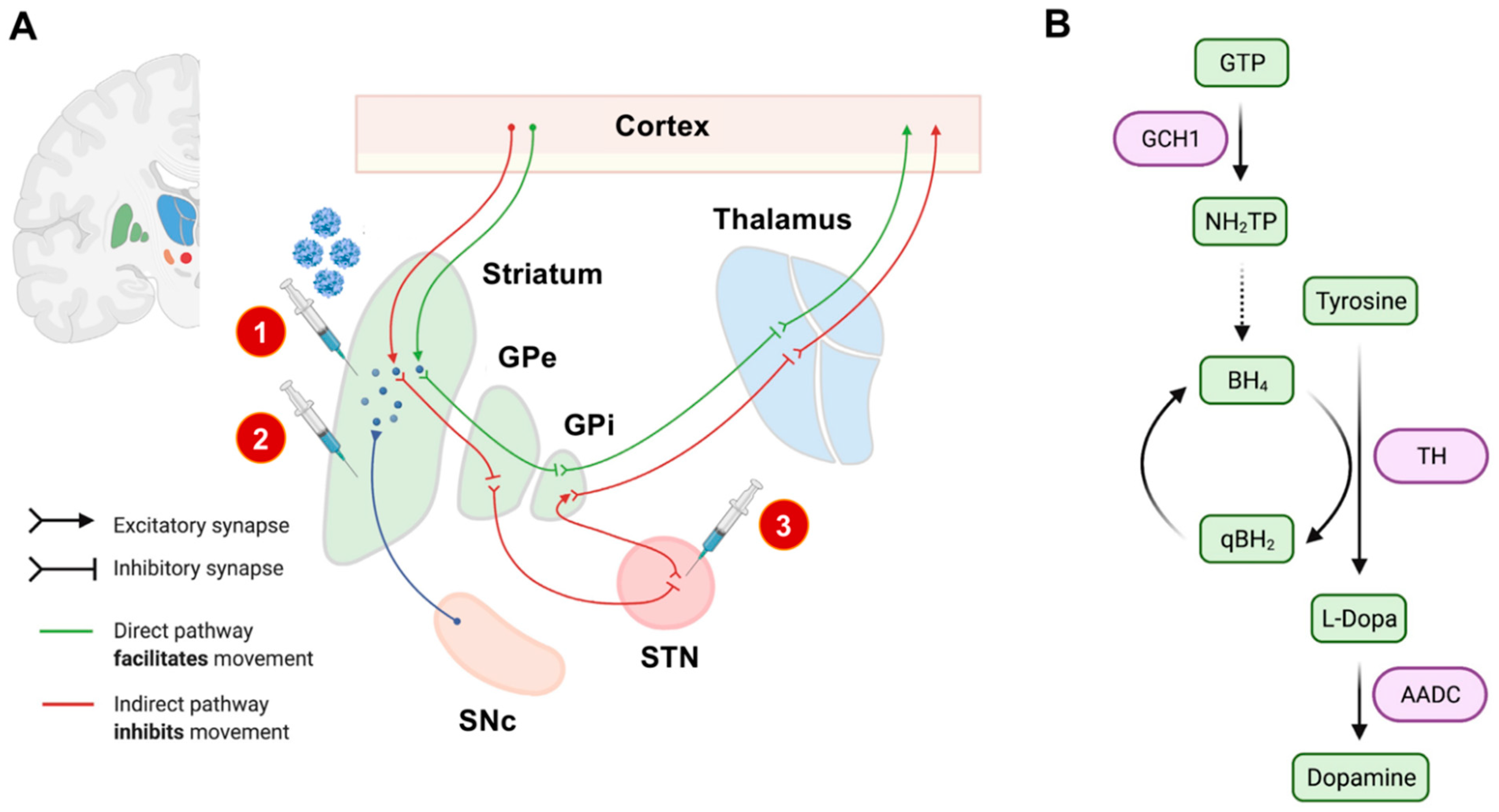
- Enhancement of dopamine synthesis by overexpression of relevant synthesis-related enzymes (tyrosinhydroxylase [TH], aromatic L-amino acid decarboxylase [AADC], GTP cyclohydrolase I [GCH1], or a combination thereof) [16].
- The overexpression of neurotrophic factors (e.g., glial cell line-derived neu-rotrophic factor [GDNF] or neurturin [NTN]) [17].
- The overexpression of glutamate decarboxylase [GAD] in the STN to decrease the synthesis of glutamate therein and to modulate basal ganglia loops in the human brain [18].
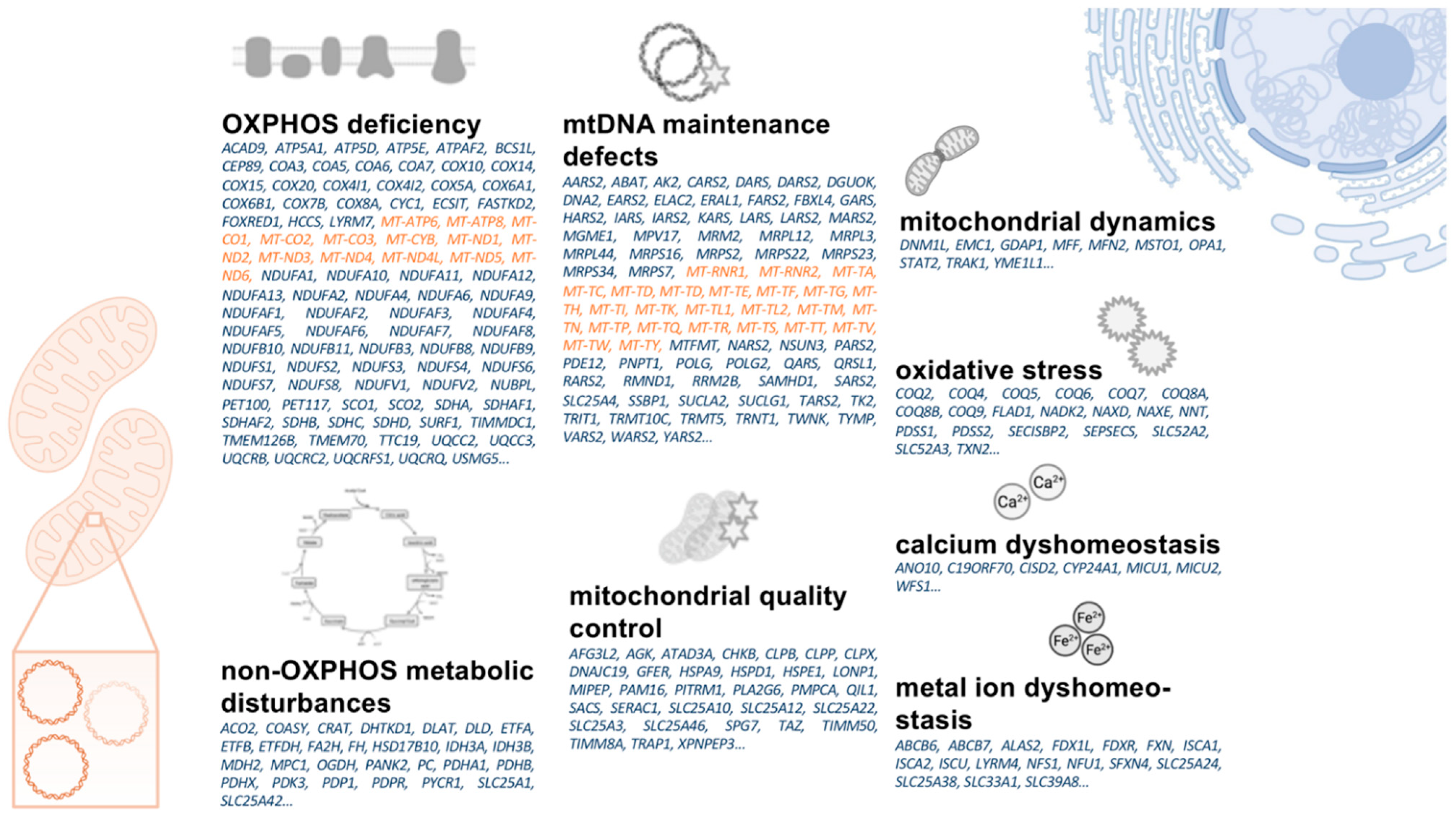
1.4. A Primer on Mitochondrial Biology
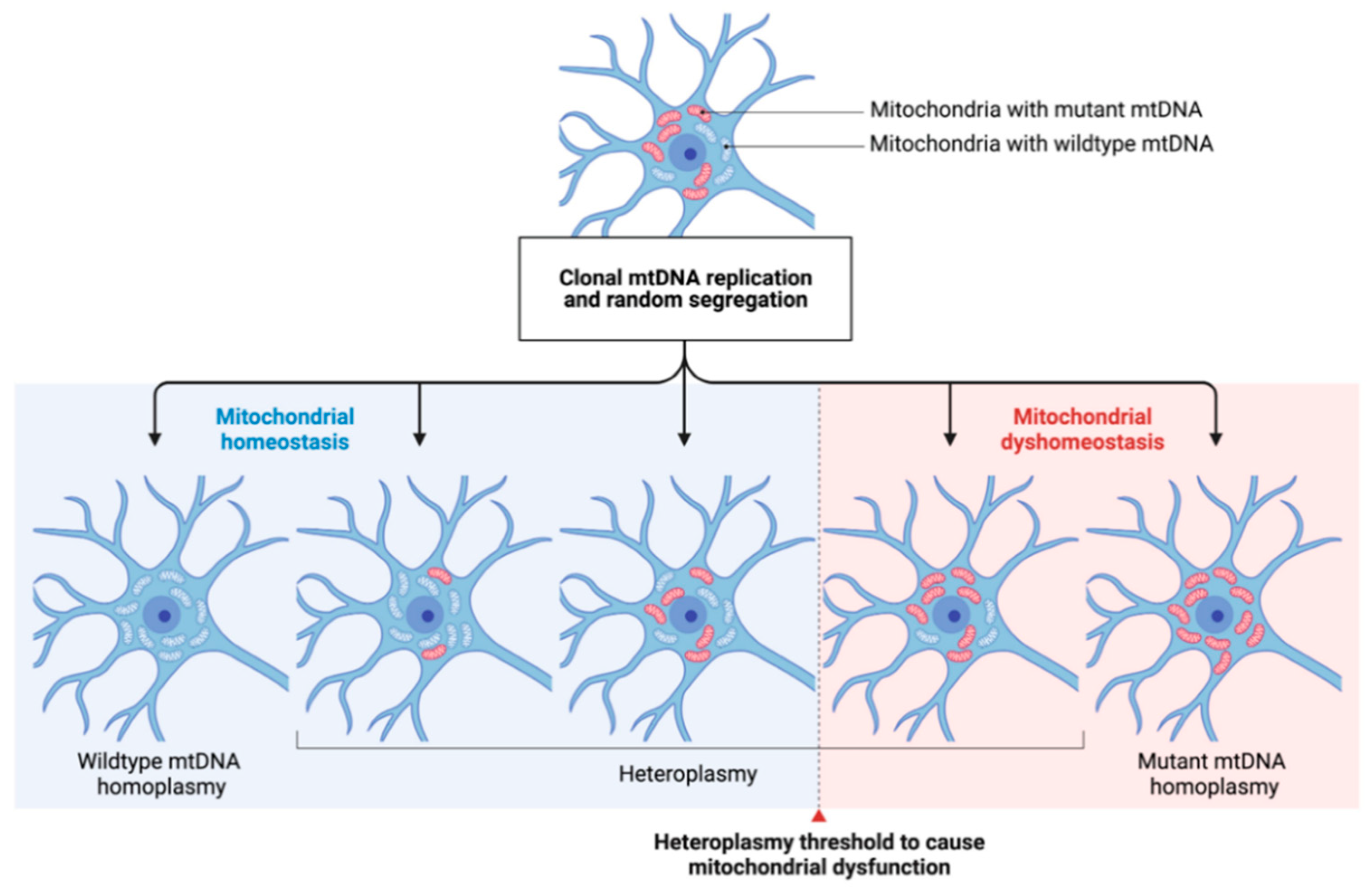
1.5. Parkinson’s Disease as a “Mitochondrial DNA Maintenance Disorder”
1.5.1. Mitochondrial DNA Changes in Aging and Neurodegeneration
- mtDNA point mutations (either inherited or somatic mutations),
- mtDNA deletions, and
- an overall reduction of mtDNA copy numbers [38].
1.5.2. Inherited and Somatic mtDNA Point Mutations and Their Role in the Pathophysiology of Parkinson’s Disease
1.5.3. The Role of Mitochondrial DNA Deletions and Copy Number Variations in PD
2. Main Body
2.1. Defining Neuroanatomical Treatment Targets
2.2. Treatment Strategies
- gene replacement/correction of monogenic PD genes,
- gene replacement of nuclear-encoded mitochondrial genes,
- allotopic expression of mtDNA-encoded genes, and
- mtDNA genome editing.
2.2.1. Gene Therapies of Monogenic Parkinson’s Disease Genes to Treat Mitochondrial Dysfunction
2.2.2. Gene Repair and Enhancement of Nuclear-Encoded Mitochondrial Genes
2.2.3. Allotopic Expression of mtDNA-Encoded Mitochondrial Genes
2.2.4. Mitochondrial DNA Genome Editing and Heteroplasmy Shifting
2.3. Special Considerations for Mitochondrial Gene Therapy
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Vijiaratnam, N.; Simuni, T.; Bandmann, O.; Morris, H.R.; Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021, 20, 559–572. [Google Scholar] [CrossRef]
- Vazquez-Velez, G.E.; Zoghbi, H.Y. Parkinson’s Disease Genetics and Pathophysiology. Annu. Rev. Neurosci. 2021, 44, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grunewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Parkinsons Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.; Israwi, A.R.; Arathoon, L.R.; Gleave, J.A.; Nash, J.E. The multi-faceted role of mitochondria in the pathology of Parkinson’s disease. J. Neurochem. 2021, 156, 715–752. [Google Scholar] [CrossRef]
- Goncalves, F.B.; Morais, V.A. PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life 2021, 11, 371. [Google Scholar] [CrossRef]
- Camilleri, A.; Vassallo, N. The centrality of mitochondria in the pathogenesis and treatment of Parkinson’s disease. CNS Neurosci. Ther. 2014, 20, 591–602. [Google Scholar] [CrossRef]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2020, 8, 615461. [Google Scholar] [CrossRef]
- Zanin, M.; Santos, B.F.R.; Antony, P.M.A.; Berenguer-Escuder, C.; Larsen, S.B.; Hanss, Z.; Barbuti, P.A.; Baumuratov, A.S.; Grossmann, D.; Capelle, C.M.; et al. Mitochondria interaction networks show altered topological patterns in Parkinson’s disease. NPJ Syst. Biol. Appl. 2020, 6, 38. [Google Scholar] [CrossRef]
- Illes, A.; Balicza, P.; Gal, A.; Pentelenyi, K.; Csaban, D.; Gezsi, A.; Molnar, V.; Molnar, M.J. Hereditary Parkinson’s disease as a new clinical manifestation of the damaged POLG gene. Orv. Hetil. 2020, 161, 821–828. [Google Scholar] [CrossRef]
- Coxhead, J.; Kurzawa-Akanbi, M.; Hussain, R.; Pyle, A.; Chinnery, P.; Hudson, G. Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol. Aging 2016, 38, 217.e1–217.e6. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Parkinson’s syndrome and Parkinson’s disease in mitochondrial disorders. Mov. Disord. 2011, 26, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Al Shahrani, M.; Heales, S.; Hargreaves, I.; Orford, M. Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease. J. Clin. Med. 2017, 6, 100. [Google Scholar] [CrossRef]
- Mohammad, R. Key considerations in formulation development for gene therapy. Drug Discov. Today 2021, in press. [Google Scholar] [CrossRef]
- Merola, A.; Van Laar, A.; Lonser, R.; Bankiewicz, K. Gene therapy for Parkinson’s disease: Contemporary practice and emerging concepts. Expert Rev. Neurother. 2020, 20, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.W.; Kiening, K.; Anselm, I.; Compton, D.R.; Nakajima, T.; Opladen, T.; Pearl, P.L.; Roubertie, A.; Roujeau, T.; Muramatsu, S.I. Gene therapy in the putamen for curing AADC deficiency and Parkinson’s disease. EMBO Mol. Med. 2021, 13, e14712. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Kumar, A.; Mehta, V.; Zengin, G.; Arora, S. Gene Therapy in the Management of Parkinson’s Disease: Potential of GDNF as a Promising Therapeutic Strategy. Curr. Gene Ther. 2020, 20, 207–222. [Google Scholar] [CrossRef]
- Niethammer, M.; Tang, C.C.; LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Sarkar, A.; et al. Long-term follow-up of a randomized AAV2-GAD gene therapy trial for Parkinson’s disease. JCI Insight 2017, 2, e90133. [Google Scholar] [CrossRef]
- Hitti, F.L.; Yang, A.I.; Gonzalez-Alegre, P.; Baltuch, G.H. Human gene therapy approaches for the treatment of Parkinson’s disease: An overview of current and completed clinical trials. Parkinsonism Relat. Disord. 2019, 66, 16–24. [Google Scholar] [CrossRef]
- Murley, A.; Nunnari, J. The Emerging Network of Mitochondria-Organelle Contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef]
- Cocco, T.; Pacelli, C.; Sgobbo, P.; Villani, G. Control of OXPHOS efficiency by complex I in brain mitochondria. Neurobiol. Aging 2009, 30, 622–629. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, W.; Kuang, X.; Hou, S.; Liu, H. Nanopreparations for mitochondria targeting drug delivery system: Current strategies and future prospective. Asian J. Pharm. Sci. 2017, 12, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Heuer, B. Mitochondrial DNA: Unraveling the “other” genome. J. Am. Assoc. Nurse Pract. 2021, 33, 673–675. [Google Scholar] [CrossRef]
- Ammal Kaidery, N.; Thomas, B. Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem. Int. 2018, 117, 91–113. [Google Scholar] [CrossRef]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef]
- Jenuth, J.P.; Peterson, A.C.; Shoubridge, E.A. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat. Genet. 1997, 16, 93–95. [Google Scholar] [CrossRef]
- Phillips, A.F.; Millet, A.R.; Tigano, M.; Dubois, S.M.; Crimmins, H.; Babin, L.; Charpentier, M.; Piganeau, M.; Brunet, E.; Sfeir, A. Single-Molecule Analysis of mtDNA Replication Uncovers the Basis of the Common Deletion. Mol. Cell 2017, 65, 527–538.e6. [Google Scholar] [CrossRef]
- Royrvik, E.C.; Johnston, I.G. MtDNA sequence features associated with ‘selfish genomes’ predict tissue-specific segregation and reversion. Nucleic Acids Res. 2020, 48, 8290–8301. [Google Scholar] [CrossRef] [PubMed]
- Kirches, E. Do mtDNA Mutations Participate in the Pathogenesis of Sporadic Parkinson’s Disease? Curr. Genom. 2009, 10, 585–593. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parakatselaki, M.E.; Ladoukakis, E.D. mtDNA Heteroplasmy: Origin, Detection, Significance, and Evolutionary Consequences. Life 2021, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Nadanaciva, S.; Murray, J.; Wilson, C.; Gebhard, D.F.; Will, Y. High-throughput assays for assessing mitochondrial dysfunction caused by compounds that impair mtDNA-encoded protein levels in eukaryotic cells. Curr. Protoc. Toxicol. 2011, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, P.; Tian, C.; O’Connell, J.; 23AndMe Research Team; Hinds, D.; Paterson, A.D.; Sondheimer, N. Nuclear genome-wide associations with mitochondrial heteroplasmy. Sci. Adv. 2021, 7, eabe7520. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Taylor, D.J.; Brown, D.T.; Manners, D.; Styles, P.; Lodi, R. Very low levels of the mtDNA A3243G mutation associated with mitochondrial dysfunction In Vivo. Ann. Neurol. 2000, 47, 381–384. [Google Scholar] [CrossRef]
- Arthur, C.R.; Morton, S.L.; Dunham, L.D.; Keeney, P.M.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol. Neurodegener. 2009, 4, 37. [Google Scholar] [CrossRef]
- Buneeva, O.; Fedchenko, V.; Kopylov, A.; Medvedev, A. Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA. Biomedicines 2020, 8, 591. [Google Scholar] [CrossRef]
- Pereira, C.V.; Gitschlag, B.L.; Patel, M.R. Cellular mechanisms of mtDNA heteroplasmy dynamics. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 510–525. [Google Scholar] [CrossRef]
- Ramon, J.; Vila-Julia, F.; Molina-Granada, D.; Molina-Berenguer, M.; Melia, M.J.; Garcia-Arumi, E.; Torres-Torronteras, J.; Camara, Y.; Marti, R. Therapy Prospects for Mitochondrial DNA Maintenance Disorders. Int. J. Mol. Sci. 2021, 22, 6447. [Google Scholar] [CrossRef]
- Kumar, R.; Harila, S.; Parambi, D.G.T.; Kanthlal, S.K.; Rahman, M.A.; Alexiou, A.; Batiha, G.E.; Mathew, B. The Role of Mitochondrial Genes in Neurodegenerative Disorders. Curr. Neuropharmacol. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Antonyova, V.; Kejik, Z.; Brogyanyi, T.; Kaplanek, R.; Pajkova, M.; Talianova, V.; Hromadka, R.; Masarik, M.; Sykora, D.; Miksatkova, L.; et al. Role of mtDNA disturbances in the pathogenesis of Alzheimer’s and Parkinson’s disease. DNA Repair 2020, 91–92, 102871. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jimenez, R.; Lurette, O.; Hebert-Chatelain, E. Damage in Mitochondrial DNA Associated with Parkinson’s Disease. DNA Cell Biol. 2020, 39, 1421–1430. [Google Scholar] [CrossRef]
- Flones, I.H.; Fernandez-Vizarra, E.; Lykouri, M.; Brakedal, B.; Skeie, G.O.; Miletic, H.; Lilleng, P.K.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; et al. Neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol. 2018, 135, 409–425. [Google Scholar] [CrossRef]
- Smigrodzki, R.; Parks, J.; Parker, W.D. High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol. Aging 2004, 25, 1273–1281. [Google Scholar] [CrossRef]
- Saha, T.; Roy, S.; Chakraborty, R.; Biswas, A.; Das, S.K.; Ray, K.; Ray, J.; Sengupta, M. Mitochondrial DNA Haplogroups and Three Independent Polymorphisms have no Association with the Risk of Parkinson’s Disease in East Indian Population. Neurol. India 2021, 69, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Li, T.; Wang, Z.F.; Huang, S.S.; Shao, Z.Q.; Wang, K.; Zhong, H.Q.; Chen, S.F.; Zhang, X.; Zhu, J.H. Mitochondrial DNA variants modulate genetic susceptibility to Parkinson’s disease in Han Chinese. Neurobiol. Dis. 2018, 114, 17–23. [Google Scholar] [CrossRef]
- Gaweda-Walerych, K.; Maruszak, A.; Safranow, K.; Bialecka, M.; Klodowska-Duda, G.; Czyzewski, K.; Slawek, J.; Rudzinska, M.; Styczynska, M.; Opala, G.; et al. Mitochondrial DNA haplogroups and subhaplogroups are associated with Parkinson’s disease risk in a Polish PD cohort. J. Neural. Transm. 2008, 115, 1521–1526. [Google Scholar] [CrossRef]
- Muller-Nedebock, A.C.; Brennan, R.R.; Venter, M.; Pienaar, I.S.; van der Westhuizen, F.H.; Elson, J.L.; Ross, O.A.; Bardien, S. The unresolved role of mitochondrial DNA in Parkinson’s disease: An overview of published studies, their limitations, and future prospects. Neurochem. Int. 2019, 129, 104495. [Google Scholar] [CrossRef]
- Oliveira, M.T.; Pontes, C.B.; Ciesielski, G.L. Roles of the mitochondrial replisome in mitochondrial DNA deletion formation. Genet Mol. Biol. 2020, 43, e20190069. [Google Scholar] [CrossRef]
- Muller-Nedebock, A.C.; van der Westhuizen, F.H.; Koks, S.; Bardien, S. Nuclear Genes Associated with Mitochondrial DNA Processes as Contributors to Parkinson’s Disease Risk. Mov. Disord. 2021, 36, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Yakubovskaya, E.; Chen, Z.; Carrodeguas, J.A.; Kisker, C.; Bogenhagen, D.F. Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J. Biol. Chem. 2006, 281, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Cantuti-Castelvetri, I.; Zheng, K.; Jackson, K.E.; Tan, Y.B.; Arzberger, T.; Lees, A.J.; Betensky, R.A.; Beal, M.F.; Simon, D.K. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann. Neurol. 2012, 71, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Bury, A.G.; Pyle, A.; Elson, J.L.; Greaves, L.; Morris, C.M.; Hudson, G.; Pienaar, I.S. Mitochondrial DNA changes in pedunculopontine cholinergic neurons in Parkinson disease. Ann. Neurol. 2017, 82, 1016–1021. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Huang, C.H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef]
- Podlesniy, P.; Puigros, M.; Serra, N.; Fernandez-Santiago, R.; Ezquerra, M.; Tolosa, E.; Trullas, R. Accumulation of mitochondrial 7S DNA in idiopathic and LRRK2 associated Parkinson’s disease. EBioMedicine 2019, 48, 554–567. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Thacker, E.A.; Toste, C.M.; Boularand, S.; Deprets, S.; Dubois, L.; Sanders, L.H. Mitochondrial DNA damage as a potential biomarker of LRRK2 kinase activity in LRRK2 Parkinson’s disease. Sci. Rep. 2020, 10, 17293. [Google Scholar] [CrossRef]
- Lujan, S.A.; Longley, M.J.; Humble, M.H.; Lavender, C.A.; Burkholder, A.; Blakely, E.L.; Alston, C.L.; Gorman, G.S.; Turnbull, D.M.; McFarland, R.; et al. Ultrasensitive deletion detection links mitochondrial DNA replication, disease, and aging. Genome Biol. 2020, 21, 248. [Google Scholar] [CrossRef]
- Langley, M.R.; Ghaisas, S.; Palanisamy, B.N.; Ay, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Characterization of nonmotor behavioral impairments and their neurochemical mechanisms in the MitoPark mouse model of progressive neurodegeneration in Parkinson’s disease. Exp. Neurol. 2021, 341, 113716. [Google Scholar] [CrossRef]
- Beckstead, M.J.; Howell, R.D. Progressive parkinsonism due to mitochondrial impairment: Lessons from the MitoPark mouse model. Exp. Neurol. 2021, 341, 113707. [Google Scholar] [CrossRef]
- Grauer, S.M.; Hodgson, R.; Hyde, L.A. MitoPark mice, an animal model of Parkinson’s disease, show enhanced prepulse inhibition of acoustic startle and no loss of gating in response to the adenosine A(2A) antagonist SCH 412348. Psychopharmacology 2014, 231, 1325–1337. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Shan, Y.; Lloyd, K.C.; Cortopassi, G.A. Mutant Twinkle increases dopaminergic neurodegeneration, mtDNA deletions and modulates Parkin expression. Hum. Mol. Genet 2012, 21, 5147–5158. [Google Scholar] [CrossRef] [PubMed]
- Dolle, C.; Flones, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Perier, C.; Bender, A.; Garcia-Arumi, E.; Melia, M.J.; Bove, J.; Laub, C.; Klopstock, T.; Elstner, M.; Mounsey, R.B.; Teismann, P.; et al. Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain 2013, 136, 2369–2378. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, Y.; Yang, Y.; Huang, X.; Sun, C. SIRT1 Protects Dopaminergic Neurons in Parkinson’s Disease Models via PGC-1alpha-Mediated Mitochondrial Biogenesis. Neurotox Res. 2021, 39, 1393–1404. [Google Scholar] [CrossRef]
- Reeve, A.K.; Grady, J.P.; Cosgrave, E.M.; Bennison, E.; Chen, C.; Hepplewhite, P.D.; Morris, C.M. Mitochondrial dysfunction within the synapses of substantia nigra neurons in Parkinson’s disease. NPJ Parkinsons Dis. 2018, 4, 9. [Google Scholar] [CrossRef]
- Vaillancourt, D.E.; Mitchell, T. Parkinson’s disease progression in the substantia nigra: Location, location, location. Brain 2020, 143, 2628–2630. [Google Scholar] [CrossRef]
- Margabandhu, G.; Vanisree, A.J. Dopamine, a key factor of mitochondrial damage and neuronal toxicity on rotenone exposure and also parkinsonic motor dysfunction-Impact of asiaticoside with a probable vesicular involvement. J. Chem. Neuroanat. 2020, 106, 101788. [Google Scholar] [CrossRef]
- Jana, S.; Sinha, M.; Chanda, D.; Roy, T.; Banerjee, K.; Munshi, S.; Patro, B.S.; Chakrabarti, S. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: Implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochem. Biophys. Acta 2011, 1812, 663–673. [Google Scholar] [CrossRef]
- Bockova, M.; Vytvarova, E.; Lamos, M.; Klimes, P.; Jurak, P.; Halamek, J.; Goldemundova, S.; Balaz, M.; Rektor, I. Cortical network organization reflects clinical response to subthalamic nucleus deep brain stimulation in Parkinson’s disease. Hum. Brain Mapp. 2021, in press. [Google Scholar] [CrossRef]
- Pinto, M.; Pickrell, A.M.; Moraes, C.T. Regional susceptibilities to mitochondrial dysfunctions in the CNS. Biol. Chem. 2012, 393, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.R. Gene therapy in the CNS-one size does not fit all. Gene Ther. 2021, 28, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Broadstock, M.; Yanez-Munoz, R.J. Challenges for gene therapy of CNS disorders and implications for Parkinson’s disease therapies. Hum. Gene Ther. 2012, 23, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Gombash, S.E.; Foust, K.D. Systemic Gene Therapy for Targeting the CNS. Methods Mol. Biol. 2016, 1382, 231–237. [Google Scholar] [CrossRef]
- Meng, Y.; Hynynen, K.; Lipsman, N. Applications of focused ultrasound in the brain: From thermoablation to drug delivery. Nat. Rev. Neurol. 2021, 17, 7–22. [Google Scholar] [CrossRef]
- Bjorklund, T.; Davidsson, M. Next-Generation Gene Therapy for Parkinson’s Disease Using Engineered Viral Vectors. J. Parkinsons Dis. 2021, 11, S209–S217. [Google Scholar] [CrossRef]
- Jayant, R.D.; Sosa, D.; Kaushik, A.; Atluri, V.; Vashist, A.; Tomitaka, A.; Nair, M. Current status of non-viral gene therapy for CNS disorders. Expert Opin. Drug Deliv. 2016, 13, 1433–1445. [Google Scholar] [CrossRef]
- Hocquemiller, M.; Giersch, L.; Audrain, M.; Parker, S.; Cartier, N. Adeno-Associated Virus-Based Gene Therapy for CNS Diseases. Hum. Gene Ther. 2016, 27, 478–496. [Google Scholar] [CrossRef]
- Safari, F.; Hatam, G.; Behbahani, A.B.; Rezaei, V.; Barekati-Mowahed, M.; Petramfar, P.; Khademi, F. CRISPR System: A High-throughput Toolbox for Research and Treatment of Parkinson’s Disease. Cell Mol. Neurobiol. 2020, 40, 477–493. [Google Scholar] [CrossRef]
- Slone, J.; Huang, T. The special considerations of gene therapy for mitochondrial diseases. NPJ Genom. Med. 2020, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Betarbet, R.; Sherer, T.B. The rotenone model of Parkinson’s disease: Genes, environment and mitochondria. Parkinsonism Relat. Disord. 2003, 9 (Suppl. S2), S59–S64. [Google Scholar] [CrossRef]
- Nicoletti, V.; Palermo, G.; Del Prete, E.; Mancuso, M.; Ceravolo, R. Understanding the Multiple Role of Mitochondria in Parkinson’s Disease and Related Disorders: Lesson from Genetics and Protein-Interaction Network. Front. Cell Dev. Biol. 2021, 9, 636506. [Google Scholar] [CrossRef] [PubMed]
- Lizama, B.N.; Chu, C.T. Neuronal autophagy and mitophagy in Parkinson’s disease. Mol. Aspects Med. 2021, 12, 100972. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Feng, S.T.; Wang, Y.T.; Yuan, Y.H.; Li, Z.P.; Chen, N.H.; Wang, Z.Z.; Zhang, Y. Mitophagy, a Form of Selective Autophagy, Plays an Essential Role in Mitochondrial Dynamics of Parkinson’s Disease. Cell Mol. Neurobiol. 2021, in press. [Google Scholar] [CrossRef]
- Buneeva, O.A.; Medvedev, A.E. DJ-1 Protein and Its Role in the Development of Parkinson’s Disease: Studies on Experimental Models. Biochemistry 2021, 86, 627–640. [Google Scholar] [CrossRef]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef]
- van der Vlag, M.; Havekes, R.; Heckman, P.R.A. The contribution of Parkin, PINK1 and DJ-1 genes to selective neuronal degeneration in Parkinson’s disease. Eur. J. Neurosci. 2020, 52, 3256–3268. [Google Scholar] [CrossRef]
- Creed, R.B.; Menalled, L.; Casey, B.; Dave, K.D.; Janssens, H.B.; Veinbergs, I.; van der Hart, M.; Rassoulpour, A.; Goldberg, M.S. Basal and Evoked Neurotransmitter Levels in Parkin, DJ-1, PINK1 and LRRK2 Knockout Rat Striatum. Neuroscience 2019, 409, 169–179. [Google Scholar] [CrossRef]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef] [PubMed]
- Risiglione, P.; Zinghirino, F.; Di Rosa, M.C.; Magri, A.; Messina, A. Alpha-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease: The Emerging Role of VDAC. Biomolecules 2021, 11, 718. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Furmston, R.; Bronstad, G.; Aasly, J.; Elliott, C.; Bandmann, O. UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2(G2019S) carriers and in vivo. Neurology 2015, 85, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Hu, Y. Mitochondrial dysfunction driven by the LRRK2-mediated pathway is associated with loss of Purkinje cells and motor coordination deficits in diabetic rat model. Cell Death Dis. 2014, 5, e1217. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Ma, P.; Liang, Q.; Yin, Y.; Wang, P.; Zhang, Q.; Wang, S.; Deng, H. Overexpression of pink1 or parkin in indirect flight muscles promotes mitochondrial proteostasis and extends lifespan in Drosophila melanogaster. PLoS ONE 2019, 14, e0225214. [Google Scholar] [CrossRef]
- Bonilla-Porras, A.R.; Arevalo-Arbelaez, A.; Alzate-Restrepo, J.F.; Velez-Pardo, C.; Jimenez-Del-Rio, M. PARKIN overexpression in human mesenchymal stromal cells from Wharton’s jelly suppresses 6-hydroxydopamine-induced apoptosis: Potential therapeutic strategy in Parkinson’s disease. Cytotherapy 2018, 20, 45–61. [Google Scholar] [CrossRef]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef]
- Liu, B.; Traini, R.; Killinger, B.; Schneider, B.; Moszczynska, A. Overexpression of parkin in the rat nigrostriatal dopamine system protects against methamphetamine neurotoxicity. Exp. Neurol. 2013, 247, 359–372. [Google Scholar] [CrossRef]
- Aguiar, A.S., Jr.; Tristao, F.S.; Amar, M.; Chevarin, C.; Lanfumey, L.; Mongeau, R.; Corti, O.; Prediger, R.D.; Raisman-Vozari, R. Parkin-knockout mice did not display increased vulnerability to intranasal administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurotox Res. 2013, 24, 280–287. [Google Scholar] [CrossRef]
- Yasuda, T.; Hayakawa, H.; Nihira, T.; Ren, Y.R.; Nakata, Y.; Nagai, M.; Hattori, N.; Miyake, K.; Takada, M.; Shimada, T.; et al. Parkin-mediated protection of dopaminergic neurons in a chronic MPTP-minipump mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2011, 70, 686–697. [Google Scholar] [CrossRef]
- Julienne, H.; Buhl, E.; Leslie, D.S.; Hodge, J.J.L. Drosophila PINK1 and parkin loss-of-function mutants display a range of non-motor Parkinson’s disease phenotypes. Neurobiol. Dis. 2017, 104, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Youle, R.J. PINK1 and Parkin flag Miro to direct mitochondrial traffic. Cell 2011, 147, 721–723. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.M.; Staveley, B.E. Expression of Pink1 with alpha-synuclein in the dopaminergic neurons of Drosophila leads to increases in both lifespan and healthspan. Genet. Mol. Res. 2012, 11, 1497–1502. [Google Scholar] [CrossRef]
- Todd, A.M.; Staveley, B.E. Pink1 suppresses alpha-synuclein-induced phenotypes in a Drosophila model of Parkinson’s disease. Genome 2008, 51, 1040–1046. [Google Scholar] [CrossRef]
- Haque, M.E.; Mount, M.P.; Safarpour, F.; Abdel-Messih, E.; Callaghan, S.; Mazerolle, C.; Kitada, T.; Slack, R.S.; Wallace, V.; Shen, J.; et al. Inactivation of Pink1 gene in vivo sensitizes dopamine-producing neurons to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and can be rescued by autosomal recessive Parkinson disease genes, Parkin or DJ-1. J. Biol. Chem. 2012, 287, 23162–23170. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Keeney, P.M.; Quigley, C.K.; Dunham, L.D.; Papageorge, C.M.; Iyer, S.; Thomas, R.R.; Schwarz, K.M.; Trimmer, P.A.; Khan, S.M.; Portell, F.R.; et al. Mitochondrial gene therapy augments mitochondrial physiology in a Parkinson’s disease cell model. Hum. Gene Ther. 2009, 20, 897–907. [Google Scholar] [CrossRef]
- Santos, D.; Cardoso, S.M. Mitochondrial dynamics and neuronal fate in Parkinson’s disease. Mitochondrion 2012, 12, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Zullo, S.J. Gene therapy of mitochondrial DNA mutations: A brief, biased history of allotopic expression in mammalian cells. Semin. Neurol. 2001, 21, 327–335. [Google Scholar] [CrossRef]
- Liufu, T.; Wang, Z. Treatment for mitochondrial diseases. Rev. Neurosci. 2021, 32, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.J.; Dixit, B.; Batiuk, E.; Hall, C.J.; O’Connor, M.S.; Boominathan, A. Codon optimization is an essential parameter for the efficient allotopic expression of mtDNA genes. Redox Biol. 2020, 30, 101429. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; McDonald, D.; Blain, A.; Sachdeva, A.; Bone, L.; Smith, A.L.M.; Warren, C.; Pickett, S.J.; Hudson, G.; Filby, A.; et al. Imaging mass cytometry reveals generalised deficiency in OXPHOS complexes in Parkinson’s disease. NPJ Parkinsons Dis. 2021, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Oca-Cossio, J.; Kenyon, L.; Hao, H.; Moraes, C.T. Limitations of allotopic expression of mitochondrial genes in mammalian cells. Genetics 2003, 165, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Martinez, F.; Vazquez-Acevedo, M.; Cortes-Hernandez, P.; Garcia-Trejo, J.J.; Davidson, E.; King, M.P.; Gonzalez-Halphen, D. What limits the allotopic expression of nucleus-encoded mitochondrial genes? The case of the chimeric Cox3 and Atp6 genes. Mitochondrion 2011, 11, 147–154. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Zullo, S.J.; Weiner, H. Factors that might affect the allotopic replacement of a damaged mitochondrial DNA-encoded protein. Rejuvenation Res. 2006, 9, 182–190. [Google Scholar] [CrossRef]
- Naeem, M.M.; Sondheimer, N. Heteroplasmy Shifting as Therapy for Mitochondrial Disorders. Adv. Exp. Med. Biol. 2019, 1158, 257–267. [Google Scholar] [CrossRef]
- Jackson, C.B.; Turnbull, D.M.; Minczuk, M.; Gammage, P.A. Therapeutic Manipulation of mtDNA Heteroplasmy: A Shifting Perspective. Trends Mol. Med. 2020, 26, 698–709. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Tanaka, M.; Borgeld, H.J.; Zhang, J.; Muramatsu, S.; Gong, J.S.; Yoneda, M.; Maruyama, W.; Naoi, M.; Ibi, T.; Sahashi, K.; et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J. Biomed. Sci. 2002, 9, 534–541. [Google Scholar] [CrossRef]
- Bacman, S.R.; Williams, S.L.; Duan, D.; Moraes, C.T. Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2012, 19, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Minczuk, M.; Papworth, M.A.; Miller, J.C.; Murphy, M.P.; Klug, A. Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 2008, 36, 3926–3938. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Bacman, S.R.; Peralta, S.; Falk, M.J.; Chomyn, A.; Chan, D.C.; Williams, S.L.; Moraes, C.T. MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases. Mol. Ther. 2015, 23, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M.; Hirano, M. Editing the Mitochondrial Genome. N. Engl. J. Med. 2020, 383, 1489–1491. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Omura, T. Mitochondria-targeting sequence, a multi-role sorting sequence recognized at all steps of protein import into mitochondria. J. Biochem. 1998, 123, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Priesnitz, C.; Becker, T. Pathways to balance mitochondrial translation and protein import. Genes Dev. 2018, 32, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017, 543, 443–446. [Google Scholar] [CrossRef]
- Mohanraj, K.; Nowicka, U.; Chacinska, A. Mitochondrial control of cellular protein homeostasis. Biochem. J. 2020, 477, 3033–3054. [Google Scholar] [CrossRef]
- Perales-Clemente, E.; Fernandez-Silva, P.; Acin-Perez, R.; Perez-Martos, A.; Enriquez, J.A. Allotopic expression of mitochondrial-encoded genes in mammals: Achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res. 2011, 39, 225–234. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Kasten, M.; Vos, M.; Konig, I.R.; Schmid, S.M.; Wilms, B.; Klein, C.; Bruggemann, N. The Use of Vitamin K2 in Patients with Parkinson’s Disease and Mitochondrial Dysfunction (PD-K2): A Theranostic Pilot Study in a Placebo-Controlled Parallel Group Design. Front. Neurol. 2020, 11, 592104. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, E.; Batista, C.; Sousa, F.; Queiroz, J.; Costa, D. Mitochondrial Gene Therapy: Advances in Mitochondrial Gene Cloning, Plasmid Production, and Nanosystems Targeted to Mitochondria. Mol. Pharm. 2017, 14, 626–638. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene Name | Mode of Inheritance | Parkinson’s Disease Phenotype | Mitochondrial Involvement in Disease Pathophysiology–Key Mechanisms | References |
|---|---|---|---|---|
| ATP13A2 (PARK9) | AR | Atypical PD, Kufor-Rakeb syndrome | Impaired mitochondrial clearance, mitochondrial dysfunction due to zinc dyshomeostasis | Ramirez et al., 2006 Grunewald et al., 2012 Park et al., 2014 |
| DJ-1 (PARK7) | AR | Early-onset PD | Reduced anti-oxidative stress mechanisms | Bonifati et al., 2003 Takahashi-Niki et al., 2004 |
| FBXO7 (PARK15) | AR | Atypical PD | Aggravated protein aggregation in mitochondria, impaired mitophagy | Shojaee et al., 2008 Zhou et al., 2018 |
| GBA | AD | resembling IPD with more rapid cognitive and motor progression, dementia with Lewy bodies | Impaired mitophagy | Sidransky et al., 2009 Barkhuizen et al., 2016 Zhao et al., 2016 Gegg et al., 2016 Moren et al., 2019 |
| LRRK2 (PARK8) | AD | resembling IPD | Disturbance in mitochondrial ATP and ROS production, impaired mitochondrial dynamics and mitophagy, mitochondrial DNA damage | Zimprich et al., 2004 Mancini et al., 2020 |
| PINK1 (PARK6) | AR | Early-onset PD | Defective mitochondrial quality control | Valente et al., 2004 Ge et al., 2020 |
| PLA2G6 (PARK14) | AR | Atypical PD, NBIA type 2B, Infantile neuroaxonal dystrophy 1 | Maintenance of mitochondrial function, impaired mitophagy | Paisan-Ruiz et al., 2009 Chiu et al., 2017 Chiu et al., 2019 |
| PRKN (PARK2) | AR | Early-onset PD | Defective mitochondrial quality control | Kitada et al., 1998 Ge et al., 2020 |
| SNCA (PARK1) | AD | May be atypical (higher frequency of cognitive/ psychiatric symptoms) | Mitochondrial toxicity, fragmented mitochondria | Polymeropoulos et al., 1997 Singleton et al., 2003 Chartier-Harlin et al., 2004 |
| VPS35 (PARK17) | AD | resembling IPD | Regulation of mitochondrial dynamics and homeostasis | Vilarino-Guell et al., 2011 Zimprich et al., 2011 Cutillo et al., 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasuhn, J.; Brüggemann, N. Gene Therapeutic Approaches for the Treatment of Mitochondrial Dysfunction in Parkinson’s Disease. Genes 2021, 12, 1840. https://doi.org/10.3390/genes12111840
Prasuhn J, Brüggemann N. Gene Therapeutic Approaches for the Treatment of Mitochondrial Dysfunction in Parkinson’s Disease. Genes. 2021; 12(11):1840. https://doi.org/10.3390/genes12111840
Chicago/Turabian StylePrasuhn, Jannik, and Norbert Brüggemann. 2021. "Gene Therapeutic Approaches for the Treatment of Mitochondrial Dysfunction in Parkinson’s Disease" Genes 12, no. 11: 1840. https://doi.org/10.3390/genes12111840
APA StylePrasuhn, J., & Brüggemann, N. (2021). Gene Therapeutic Approaches for the Treatment of Mitochondrial Dysfunction in Parkinson’s Disease. Genes, 12(11), 1840. https://doi.org/10.3390/genes12111840