
The Genetic Landscape of Inherited Retinal Diseases in a Mexican Cohort: Genes, Mutations and Phenotypes

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
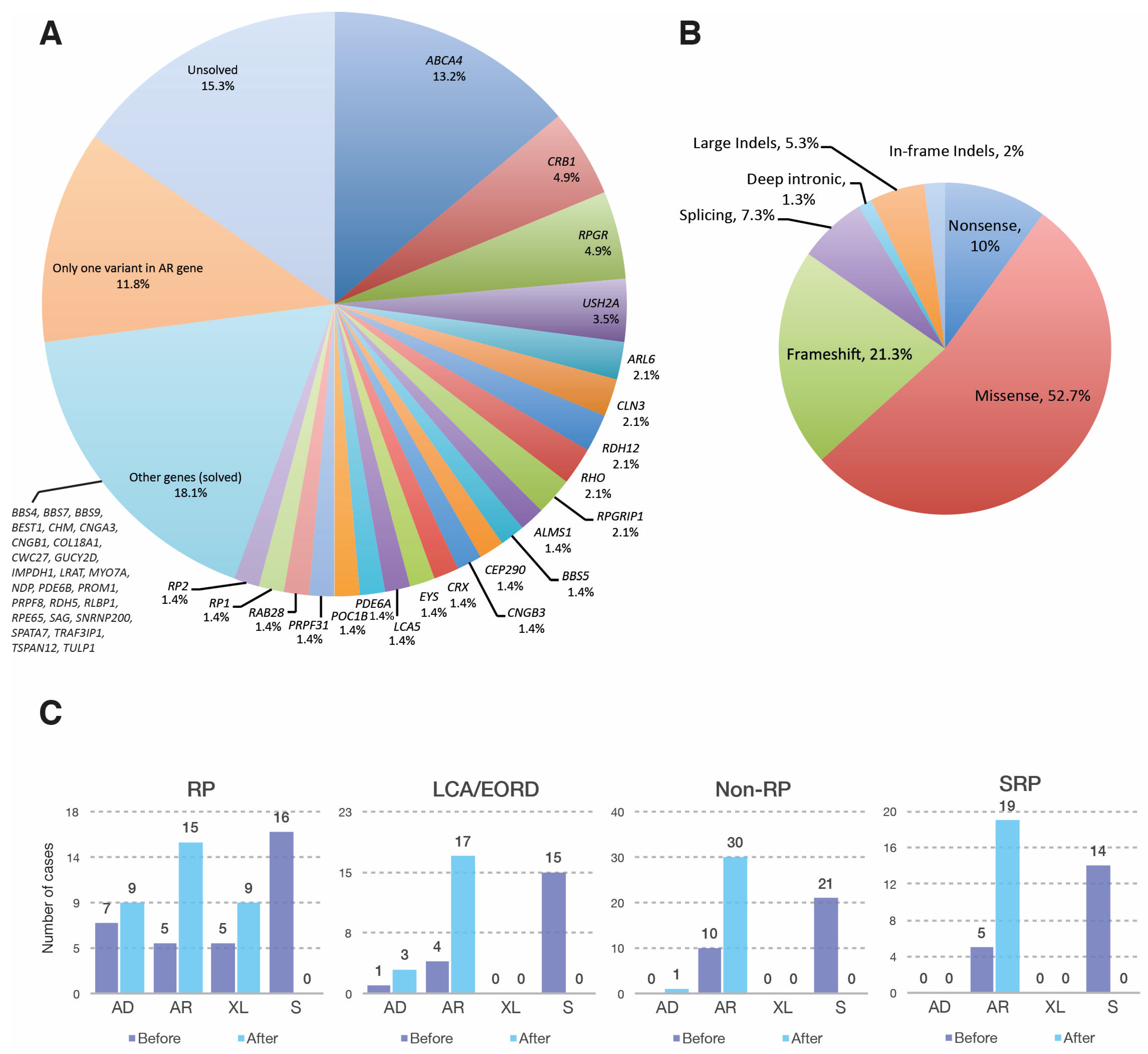
3.1. Genetic Landscape of the Mexican IRD Patients
3.1.1. Retinitis Pigmentosa
3.1.2. Leber Congenital Amaurosis and Early Onset Retinal Dystrophy
3.1.3. Non-RP Retinal Dystrophies
3.1.4. Syndromic Retinal Dystrophies
3.1.5. Other Retinal Dystrophies
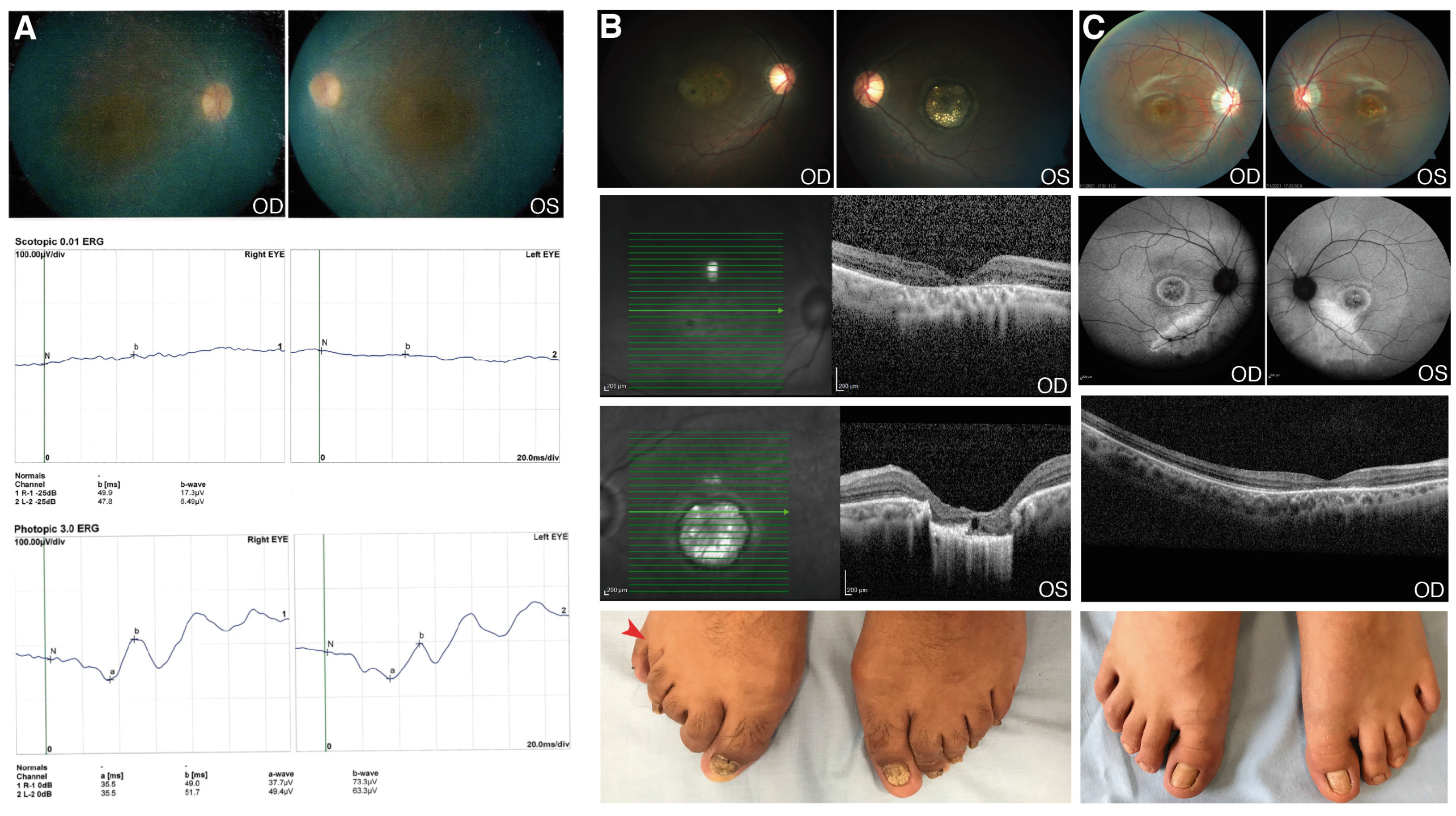
3.2. Novel Genetic Findings in Rare IRD Cases
3.2.1. CWC27
3.2.2. RAB28
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef]
- Solebo, A.L.; Teoh, L.; Rahi, J. Epidemiology of blindness in children. Arch. Dis. Child. 2017, 102, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef] [PubMed]
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Tatour, Y.; Ben-Yosef, T. Syndromic inherited retinal diseases: Genetic, clinical and diagnostic aspects. Diagnostics 2020, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Gonzàlez-Duarte, R.; de Castro-Miró, M.; Tuson, M.; Ramírez-Castañeda, V.; Valero Gils, R.; Marfany, G. Scaling New Heights in the Genetic Diagnosis of Inherited Retinal Dystrophies. Adv. Exp. Med. Biol. 2019, 1185, 215–219. [Google Scholar]
- Toulis, V.; Cortés-González, V.; Castro-Miró, M.; Sallum, J.F.; Català-Mora, J.; Villanueva-Mendoza, C.; Ciccioli, M.; Gonzàlez-Duarte, R.; Valero, R.; Marfany, G. Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants. Genes 2020, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-F.; Huang, F.; Wu, K.-C.; Wu, J.; Chen, J.; Pang, C.-P.; Lu, F.; Qu, J.; Jin, Z.-B. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hosono, K.; Nishina, S.; Yokoi, T.; Katagiri, S.; Saitsu, H.; Kurata, K.; Miyamichi, D.; Hikoya, A.; Mizobuchi, K.; Nakano, T.; et al. Molecular diagnosis of 34 Japanese families with leber congenital amaurosis using targeted next generation sequencing. Sci. Rep. 2018, 8, 8279. [Google Scholar] [CrossRef]
- Ku, C.; Hull, S.; Arno, G.; Vincent, A.; Carss, K.; Kayton, R.; Weeks, D.; Anderson, G.; Geraets, R.; Parker, C.; et al. Detailed Clinical Phenotype and Molecular Genetic Findings in CLN3-Associated Isolated Retinal Degeneration. JAMA Ophthalmol. 2017, 135, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, H.; Tuan, H.; Nguyen, D.; Sun, V.; Keser, V.; Bowne, S.; Sullivan, L.; Luo, H.; Zhao, L.; et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 2014, 133, 331–345. [Google Scholar] [CrossRef]
- Zenteno, J.C.; García-Montaño, L.A.; Cruz-Aguilar, M.; Ronquillo, J.; Rodas-Serrano, A.; Aguilar-Castul, L.; Matsui, R.; Vencedor-Meraz, C.I.; Arce-González, R.; Graue-Wiechers, F.; et al. Extensive genic and allelic heterogeneity underlying inherited retinal dystrophies in Mexican patients molecularly analyzed by next-generation sequencing. Mol. Genet. Genomic Med. 2020, 8, e1044. [Google Scholar] [CrossRef] [PubMed]
- Talib, M.; Van Cauwenbergh, C.; De Zaeytijd, J.; Van Wynsberghe, D.; De Baere, E.; Boon, C.J.F.; Leroy, B.P. CRB1-associated retinal dystrophies in a Belgian cohort: Genetic characteristics and long-term clinical follow-up. Br. J. Ophthalmol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Aldahmesh, M.A.; Mohamed, J.Y.; Al-Mesfer, S.; Alkuraya, F.S. The distinct ophthalmic phenotype of Knobloch syndrome in children. Br. J. Ophthalmol. 2012, 96, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xie, Y.A.; Abouzeid, H.; Gordon, C.T.; Fiorentino, A.; Sun, Z.; Lehman, A.; Osman, I.S.; Dharmat, R.; Riveiro-Alvarez, R.; et al. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am. J. Hum. Genet. 2017, 100, 592–604. [Google Scholar] [CrossRef]
- Jespersgaard, C.; Hey, A.B.; Ilginis, T.; Hjortshøj, T.D.; Fang, M.; Bertelsen, M.; Bech, N.; Jensen, H.; Larsen, L.J.; Tümer, Z.; et al. A missense mutation in Rab28 in a family with cone-rod dystrophy and postaxial polydactyly prevents localization of Rab28 to the primary cilium. Investig. Ophthalmol. Vis. Sci. 2020, 61, 29. [Google Scholar] [CrossRef]
- Brea-Fernández, A.J.; Cabanas, P.; Dacruz-Álvarez, D.; Caamaño, P.; Limeres, J.; Loidi, L. Expanding the clinical and molecular spectrum of the CWC27-related spliceosomopathy. J. Hum. Genet. 2019, 64, 1133–1136. [Google Scholar] [CrossRef]
- Iarossi, G.; Marino, V.; Maltese, P.E.; Colombo, L.; D’Esposito, F.; Manara, E.; Dhuli, K.; Modarelli, A.M.; Cennamo, G.; Magli, A.; et al. Expanding the Clinical and Genetic Spectrum of RAB28-Related Cone-Rod Dystrophy: Pathogenicity of Novel Variants in Italian Families. Int. J. Mol. Sci. 2021, 22, 381. [Google Scholar] [CrossRef]
- Lee, G.I.; Lee, C.; Subramanian, S.; Kim, N.K.D.; Ki, C.S.; Park, W.Y.; Kim, B.J.; Kim, S.J. A novel likely pathogenic variant in the RAB28 gene in a Korean patient with cone–rod dystrophy. Ophthalmic Genet. 2017, 38, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Álvarez, R.; Xie, Y.; López-Martínez, M.Á.; Gambin, T.; Pérez-Carro, R.; Ávila-Fernández, A.; López-Molina, M.I.; Zernant, J.; Jhangiani, S.; Muzny, D.; et al. New mutations in the RAB28 gene in 2 spanish families with conerod dystrophy. JAMA Ophthalmol. 2015, 133, 133–139. [Google Scholar] [CrossRef][Green Version]
- Roosing, S.; Rohrschneider, K.; Beryozkin, A.; Sharon, D.; Weisschuh, N.; Staller, J.; Kohl, S.; Zelinger, L.; Peters, T.A.; Neveling, K.; et al. Mutations in RAB28, encoding a farnesylated small gtpase, are associated with autosomal-recessive cone-rod dystrophy. Am. J. Hum. Genet. 2013, 93, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, Y.; Akiyama, M.; Nishiguchi, K.M.; Momozawa, Y.; Kamatani, Y.; Takata, S.; Inai, C.; Iwasaki, Y.; Kumano, M.; Murakami, Y.; et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. J. Med. Genet. 2019, 56, 662–670. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, Y.N.; Yoon, Y.H.; Seo, E.J.; Seo, G.H.; Keum, C.; Lee, B.H.; Lee, J.Y. Diverse genetic landscape of suspected retinitis pigmentosa in a large korean cohort. Genes 2021, 12, 675. [Google Scholar] [CrossRef] [PubMed]
- Remmer, M.H.; Rastogi, N.; Ranka, M.P.; Ceisler, E.J. Achromatopsia: A review. Curr. Opin. Ophthalmol. 2015, 26, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Hinman, E.G.; Collin, G.B.; Beck, S.; Cerqueira, R.; Maffei, P.; Milan, G.; Zhang, W.; Wilson, D.I.; Hearn, T.; et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum. Mutat. 2007, 28, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Camacho, O.F.; Zenteno, J.C. Review and update on the molecular basis of Leber congenital amaurosis. World J. Clin. Cases WJCC 2015, 3, 112. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guan, L.; Xiao, X.; Zhang, J.; Li, S.; Jiang, H.; Jia, X.; Yin, Y.; Guo, X.; Wang, J.; et al. ALMS1 null mutations: A common cause of Leber congenital amaurosis and early-onset severe cone-rod dystrophy. Clin. Genet. 2016, 89, 442–447. [Google Scholar] [CrossRef]
- Busetto, V.; Barbosa, I.; Basquin, J.; Marquenet, É.; Hocq, R.; Hennion, M.; Paternina, J.A.; Namane, A.; Conti, E.; Bensaude, O.; et al. Structural and functional insights into CWC27/CWC22 heterodimer linking the exon junction complex to spliceosomes. Nucleic Acids Res. 2020, 48, 5670. [Google Scholar] [CrossRef] [PubMed]
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab family of small GTPases: An updated view on their regulation and functions. FEBS J. 2021, 288, 36–55. [Google Scholar] [CrossRef] [PubMed]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R4860. [Google Scholar] [CrossRef] [PubMed]
- Nassari, S.; Del Olmo, T.; Jean, S. Rabs in Signaling and Embryonic Development. Int. J. Mol. Sci. 2020, 21, 1064. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.; Boldt, K.; Ueffing, M.; Gerstner, C.D.; Frederick, J.M.; Baehr, W. The small GTPase RAB28 is required for phagocytosis of cone outer segments by the murine retinal pigmented epithelium. J. Biol. Chem. 2018, 293, 17546–17558. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Case | Gene | NM Number | Chr | HGVS DNA | HGVS Protein Change | Zyg | Inh | ACMG |
|---|---|---|---|---|---|---|---|---|
| 5 | ABCA4 | NM_000350.3 | 1 | c.3415T>G | p.(Tyr1139Asp) | Het | AR | VUS |
| 19 | ABCA4 | NM_000350.3 | 1 | c.6299G>A; c.6308C>A | p.(Gly2100Glu); p.(Pro2103His) | Het | AR | LPV; LPV |
| 21 | ALMS1 | NM_015120.4 | 2 | c.6828C>A | p.(Cys2276Ter) | Hom | AR | LPV |
| 20 | ALMS1 | NM_015120.4 | 2 | c.7881_7882insGACA | p.(Leu2628AspfsTer33) | Hom | AR | LPV |
| 23 | ARL6 | NM_177976.3 | 3 | c.482C>T | p.(Ala161Val) | Het | AR | VUS |
| 25 | BBS4 | NM_033028.5 | 15 | c.187C>T | p.(Gln63Ter) | Hom | AR | PV |
| 26 | BBS5 | NM_152384.3 | 2 | c.143-1G>C | Hom | AR | PV | |
| 27 | BBS5 | NM_152384.3 | 2 | c.143-1G>C | Het | AR | PV | |
| 27 | BBS5 | NM_152384.3 | 2 | c.559_560insGA | p.(Ile187ArgfsTer8) | Het | AR | PV |
| 28 | BBS7 | NM_176824.3 | 4 | c.302T>A | p.(Leu101His) | Hom | AR | VUS |
| 35 | CLN3 | NM_001042432.2 | 16 | c.147C>G | p.(Asn49Lys) | Hom | AR | VUS |
| 40 | CNGB3 | NM_019098.5 | 8 | c.701G>A | p.(Cys234Tyr) | Het | AR | VUS |
| 42 | CRB1 | NM_201253.3 | 1 | c.2630_2631dup | p.(Leu878PhefsTer5) | Het | AR | PV |
| 43 | CRB1 | NM_201253.3 | 1 | c.3166G>T | p.(Asp1056Tyr) | Hom | AR | LPV |
| 45 | CRB1 | NM_201253.3 | 1 | c.3881G>A | p.(Cys1294Tyr) | Het | AR | LPV |
| 46 | CRB1 | NM_201253.3 | 1 | c.3884_3904del | p.(Glu1295_Cys1301del) | Het | AR | LPV |
| 47 | CRB1 | NM_201253.3 | 1 | Deletion of exons 8-9 and insertion of exon 6 (inverted) | Hom | AR | PV | |
| 50 | CRX | NM_000554.6 | 19 | c.564del | p.(Ala189ProfsTer5) | Het | AD | PV |
| 51 | CWC27 | NM_005869.4 | 5 | c.1066_1070del | p.(Ala356CysfsTer11) | Hom | AR | PV |
| 53 | EYS | NM_001142800.2 | 6 | c.2287T>G | p.(Trp763Gly) | Het | AR | VUS |
| 53 | EYS | NM_001142800.2 | 6 | c.8606C>G | p.(Ser2869Ter) | Het | AR | PV |
| 52 | EYS | NM_001142800.2 | 6 | Duplication of exons 4-5 | Het | AR | PV | |
| 54 | GUCY2D | NM_000180.4 | 17 | c.1773del | p.(Asn591LysfsTer46) | Het | AR | PV |
| 55 | IMPDH1 | NM_000883.4 | 7 | c.940A>G | p.(Lys314Glu) | Het | AD | VUS |
| 56 | LCA5 | NM_001122769.3 | 6 | c.66_72delTTACTTins302nt | Hom | AR | PV | |
| 57 | LCA5 | NM_001122769.3 | 6 | c.1368dup | p.(Glu457ArgfsTer14) | Hom | AR | PV |
| 58 | LRAT | NM_004744.5 | 4 | c.224C>T | p.(Pro75Leu) | Het | AR | LPV |
| 58 | LRAT | NM_004744.5 | 4 | c.504C>A | p.(Cys168Ter) | Het | AR | PV |
| 59 | MYO7A | NM_000260.4 | 11 | c.767A>G | p.(Tyr256Cys) | Het | AR | LPV |
| 59 | MYO7A | NM_000260.4 | 11 | c.6071G>C | p.(Arg2024Pro) | Het | AR | LPV |
| 60 | NDP | NM_000266.4 | X | c.355A>C | p.(Thr119Pro) | Hem | XL | LPV |
| 61 | PDE6A | NM_000440.3 | 5 | c.2380_2382del | p.(Glu794del) | Het | AR | LPV |
| 63 | PDE6B | NM_000283.4 | 4 | c.1682A>G | p.(His561Arg) | Hom | AR | LPV |
| 65 | POC1B | NM_172240.3 | 12 | c.144del | p.(Lys48AsnfsTer16) | Het | AR | PV |
| 67 | PRPF31 | NM_015629.4 | 19 | c.176del | p.(Met59SerfsTer6) | Het | AD | PV |
| 70, 71 | RAB28 | NM_001017979.3 | 4 | c.202G>C | p.(Asp68His) | Hom | AR | VUS |
| 72 | RDH12 | NM_152443.3 | 14 | c.529G>C | p.(Ala177Pro) | Hom | AR | VUS |
| 79 | RLBP1 | NM_000326.5 | 15 | Deletion of exon 6 | Hom | AR | PV | |
| 81 | RP1 | NM_006269.2 | 8 | c.4709del | p.(Gly1570GlufsTer10) | Het | AR | LPV |
| 83 | RP2 | NM_006915.3 | X | c.524A>C | p.(His175Pro) | Hem | XL | LPV |
| 87 | RPGR | NM_001034853.2 | X | c.736_745dup | p.(Ala249AspfsTer37) | Hem | XL | PV |
| 88 | RPGR | NM_001034853.2 | X | c.1481G>T | p.(Gly494Val) | Hem | XL | VUS |
| 89 | RPGR | NM_001034853.2 | X | c.2455_2468dup | Hem | XL | VUS | |
| 90 | RPGR | NM_001034853.2 | X | c.2543del | p.(Glu848GlyfsTer241) | Hem | XL | PV |
| 91 | RPGR | NM_001034853.2 | X | c.2587G>T | p.(Glu863Ter) | Hem | XL | VUS |
| 93 | RPGRIP1 | NM_020366.4 | 14 | c.2988del | p.(Glu996AspfsTer5) | Het | AR | LPV |
| 93 | RPGRIP1 | NM_020366.4 | 14 | Deletion of exons 2–17 | Het | AR | PV | |
| 98 | TRAF3IP1 | NM_015650.4 | 2 | c.88C>T | p.(Pro30Ser) | Hom | AR | VUS |
| 99 | TSPAN12 | NM_012338.4 | 7 | c.301dup | p.(Leu101ProfsTer16) | Het | AD | PV |
| 102 | USH2A | NM_206933.4 | 1 | c.9473del | p.(Lys3158SerfsTer2) | Het | AR | PV |
| 105 | USH2A | NM_206933.4 | 1 | c.8126_8127dupCA | p.(Asn2710GlnfsTer7) | Hom | AR | PV |
| IRD | Patients | Genes Identified |
|---|---|---|
| Non-syndromic | ||
| Achromatopsia | 3 | CNGA3 (1-AR), CNGB3 (2-AR) |
| Best disease | 1 | BEST1 (1-AD) |
| Choroideremia | 1 | CHM (1-XL) |
| Cone-rod dystrophy/cone dystrophy | 8 | ABCA4 (1-AR), ARL6 (1-AR), CRB1 (1-AR), POC1B (2-AR), PROM1 (1-AR), RAB28 (1-AR), RPGRIP1 (1-AR) |
| Congenital stationary night blindness | 1 | RDH5 (1-AR) |
| Early-onset retinal dystrophy | 6 | CEP290 (1-AR), CRB1 (1-AR), CWC27 (1-AR), GUCY2D (1-AR), RDH12 (2-AR) |
| Familial exudative vitreoretinopathy | 1 | TSPAN12 (1-AD) |
| Leber congenital amaurosis | 14 | CEP290 (1-AR), CRB1 (4-AR), CRX (2-AD), IMPDH1 (1-AD), LCA5 (2-AR), RPE65 (1-AR), RPGRIP1 (2-AR), SPATA7 (1-AR) |
| Maculopathy, retinal degeneration | 1 | CLN3 (1-AR) |
| Norrie disease | 1 | NDP (1-XL) |
| Retinitis pigmentosa | 32 | ABCA4 (1-AR), CNGB1 (1-AR), CRB1 (1-AR), EYS (2-AR), LRAT (1-AR), PDE6A (2-AR), PDE6B (1-AR), PRPF31 (2-AD), PRPF8 (1-AD), RDH12 (1-AR), RHO (3-AD), RLBP1 (1-AR), RP1 (1-AD, 1-AR), RP2 (2-XL), RPGR (7-XL), SAG (1-AD), SNRNP200 (1-AD), TULP1 (1-AR), USH2A (1-AR) |
| Stargardt disease | 17 | ABCA4 (17-AR) |
| Syndromic | ||
| Alström syndrome | 2 | ALMS1 (2-AR) |
| Bardet-Biedl syndrome | 7 | ARL6 (2-AR), BBS4 (1-AR), BBS5 (2-AR), BBS7 (1-AR), BBS9 (1-AR) |
| Con-rod dystrophy, syndromic | 1 | RAB28 (1-AR) |
| Batten disease/JNCL | 2 | CLN3 (2-AR) |
| Knobloch syndrome | 1 | COL18A1 (1-AR) |
| Senior-Løken syndrome | 1 | TRAF3IP1 (1-AR) |
| Usher syndrome | 5 | USH2A (4-AR), MYO7A (1-AR) |
| Total | 105 |
| Case | Initial Diagnosis | Gene | Final Diagnosis |
|---|---|---|---|
| 3 | Maculopathy | ABCA4 | STGD |
| 35 | STGD | CLN3 | Maculopathy and RD |
| 48 | STGD | CRB1 | CRD |
| 43 | RP vs. STGD | CRB1 | RP |
| 33 | RP | CHM | Choroideremia |
| 103 | RP | USH2A | Usher II |
| 38 | ACHR vs. BCM | CNGA3 | ACHR |
| 65 | ACHR | POC1B | CRD |
| 94 | ACHR | RPGRIP1 | CRD |
| 20 | LCA | ALMS1 | Alström syndrome |
| 21 | LCA | ALMS1 | Alström syndrome |
| 87 | EORD | RPGR | XLRP |
| 51 | EORD vs. XLRP | CWC27 | EORD |
| 36 | EORD | CLN3 | Protracted JNCL |
| 28 | BBS vs. Alström | BBS7 | BBS |
| 41 | SRP | COL18A | Knobloch syndrome |
| 70 | SRP | RAB28 | CRD + polydactyly |
| 104 | Usher II vs. Usher III | USH2A | Usher II |
| Gene | HGVS DNA | HGVS Protein Change | Phe | Zyg | Ref |
|---|---|---|---|---|---|
| CWC27 | c.19C>T | p.(Gln7Ter) | RP, syndromic | Het | [17] |
| c.355C>T | p.(Arg119Ter) | RP, syndromic | Hom | [19] | |
| c.427C>T | p.(Arg143Ter) | RP, syndromic | Het | [17] | |
| c.495G>A | p.(Leu167GlyfsTer3) | RP, syndromic | Hom | [17] | |
| c.599+1G>A | p.[Val166LysfsTer3; Val191LysfsTer3] | RP, syndromic | Hom | [17] | |
| c.617C> | p.(Ser206Ter) | RD | Het | [17] | |
| c.943G>T | p.(Glu315Ter) | RP, syndromic | Hom | [17] | |
| c.1002dupA | p.(Val335SerfsTer13) | RP | Het | [17] | |
| c.1066_1070delGCTGT | p.(Ala356CysfsTer11) | EORD | Hom | This study | |
| RAB28 | c.37del | p.(Leu13Ter) | CRD (AR) | Hom | [20] |
| c.55G>A | p.(Gly19Arg) | CRD (AR), PAP, and myopia | Hom | [18] | |
| c.68C>T | p.(Ser23Phe) | CRD (AR) | Hom | [21] | |
| c.76−9A>G | p.(Thr26ValfsTer4) | CRD (AR) | Hom | [20] | |
| c.77C>A | p.(Thr26Asn) | CRD (AR) | Het | [20] | |
| c.172+1G>C | CRD (AR) | Hom | [22] | ||
| c.202G>C | p.(Asp68His) | CRD (AR) with or without unilateral PAP | Hom | This study | |
| c.321G>A | p.(Trp107Ter) | CRD (AR) | Hom | [20] | |
| c.409C>T | p.(Arg137Ter) | CRD (AR) | Hom/Het | [20,23] | |
| c.565C>T | p.(Gln189Ter) | CRD (AR) | Hom | [23] | |
| c.651T>G | p.(Cys217Trp) | CRD (AR) | Hom | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villanueva-Mendoza, C.; Tuson, M.; Apam-Garduño, D.; de Castro-Miró, M.; Tonda, R.; Trotta, J.R.; Marfany, G.; Valero, R.; Cortés-González, V.; Gonzàlez-Duarte, R. The Genetic Landscape of Inherited Retinal Diseases in a Mexican Cohort: Genes, Mutations and Phenotypes. Genes 2021, 12, 1824. https://doi.org/10.3390/genes12111824
Villanueva-Mendoza C, Tuson M, Apam-Garduño D, de Castro-Miró M, Tonda R, Trotta JR, Marfany G, Valero R, Cortés-González V, Gonzàlez-Duarte R. The Genetic Landscape of Inherited Retinal Diseases in a Mexican Cohort: Genes, Mutations and Phenotypes. Genes. 2021; 12(11):1824. https://doi.org/10.3390/genes12111824
Chicago/Turabian StyleVillanueva-Mendoza, Cristina, Miquel Tuson, David Apam-Garduño, Marta de Castro-Miró, Raul Tonda, Jean Remi Trotta, Gemma Marfany, Rebeca Valero, Vianney Cortés-González, and Roser Gonzàlez-Duarte. 2021. "The Genetic Landscape of Inherited Retinal Diseases in a Mexican Cohort: Genes, Mutations and Phenotypes" Genes 12, no. 11: 1824. https://doi.org/10.3390/genes12111824
APA StyleVillanueva-Mendoza, C., Tuson, M., Apam-Garduño, D., de Castro-Miró, M., Tonda, R., Trotta, J. R., Marfany, G., Valero, R., Cortés-González, V., & Gonzàlez-Duarte, R. (2021). The Genetic Landscape of Inherited Retinal Diseases in a Mexican Cohort: Genes, Mutations and Phenotypes. Genes, 12(11), 1824. https://doi.org/10.3390/genes12111824