
Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population

Abstract
1. Introduction
2. Materials and Methods
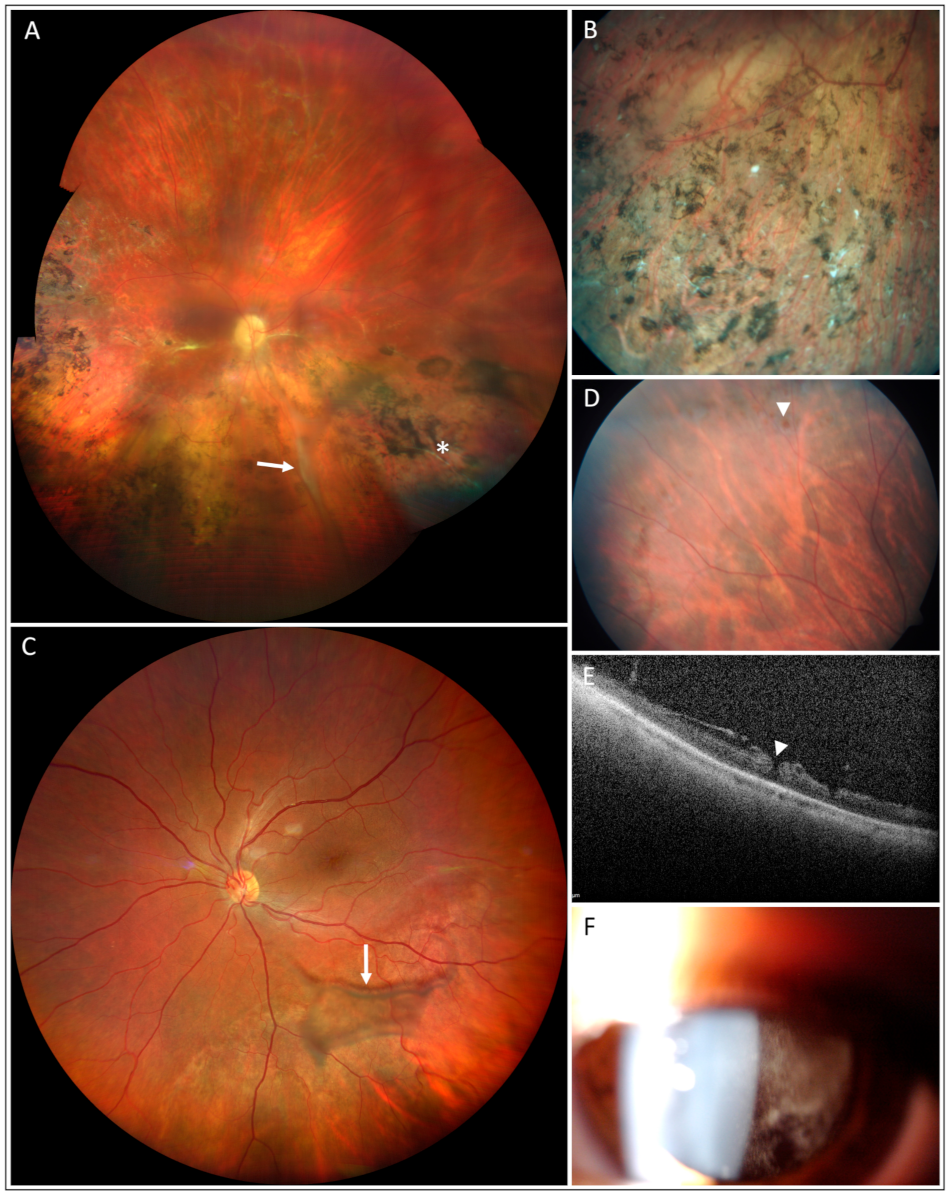
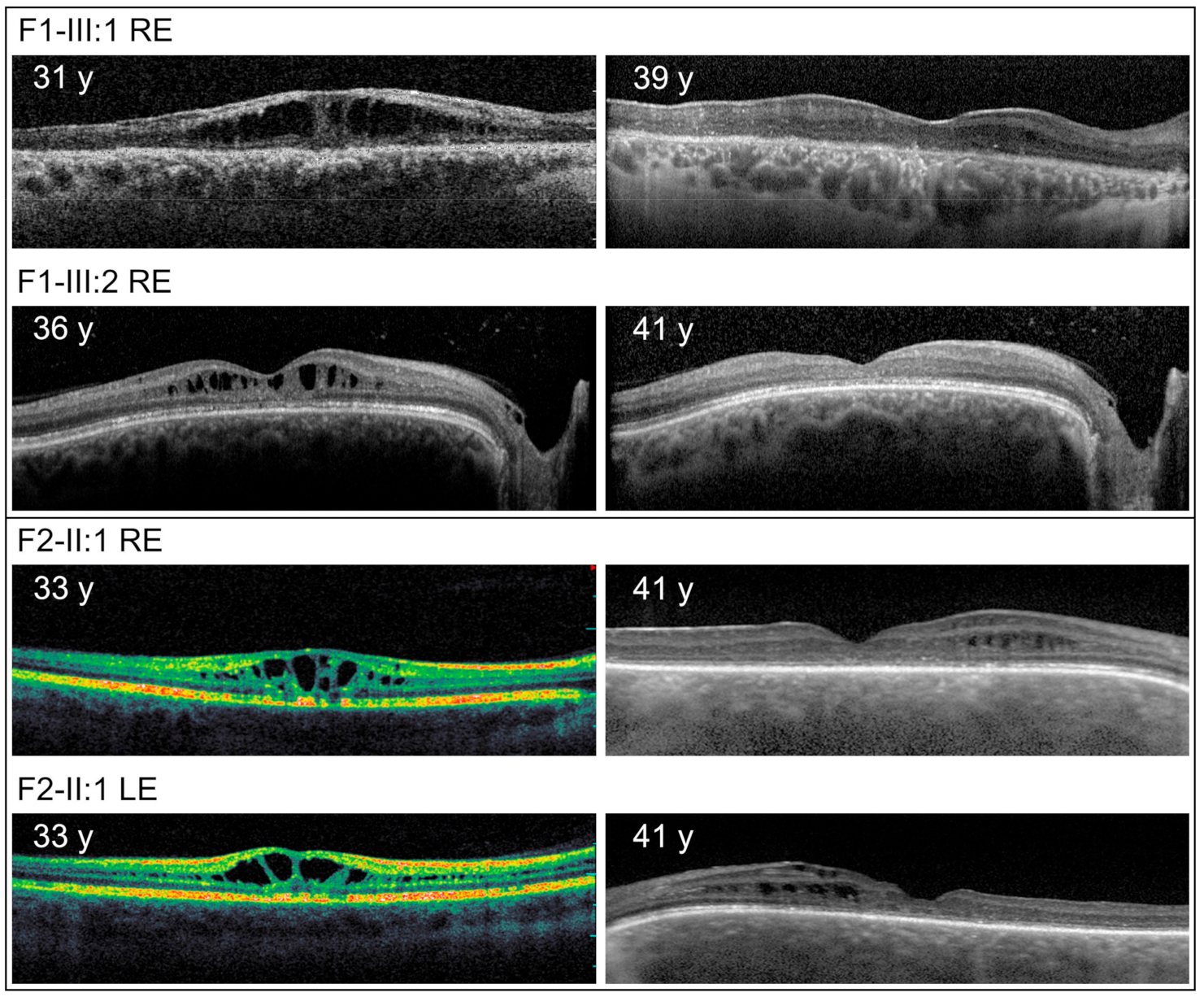
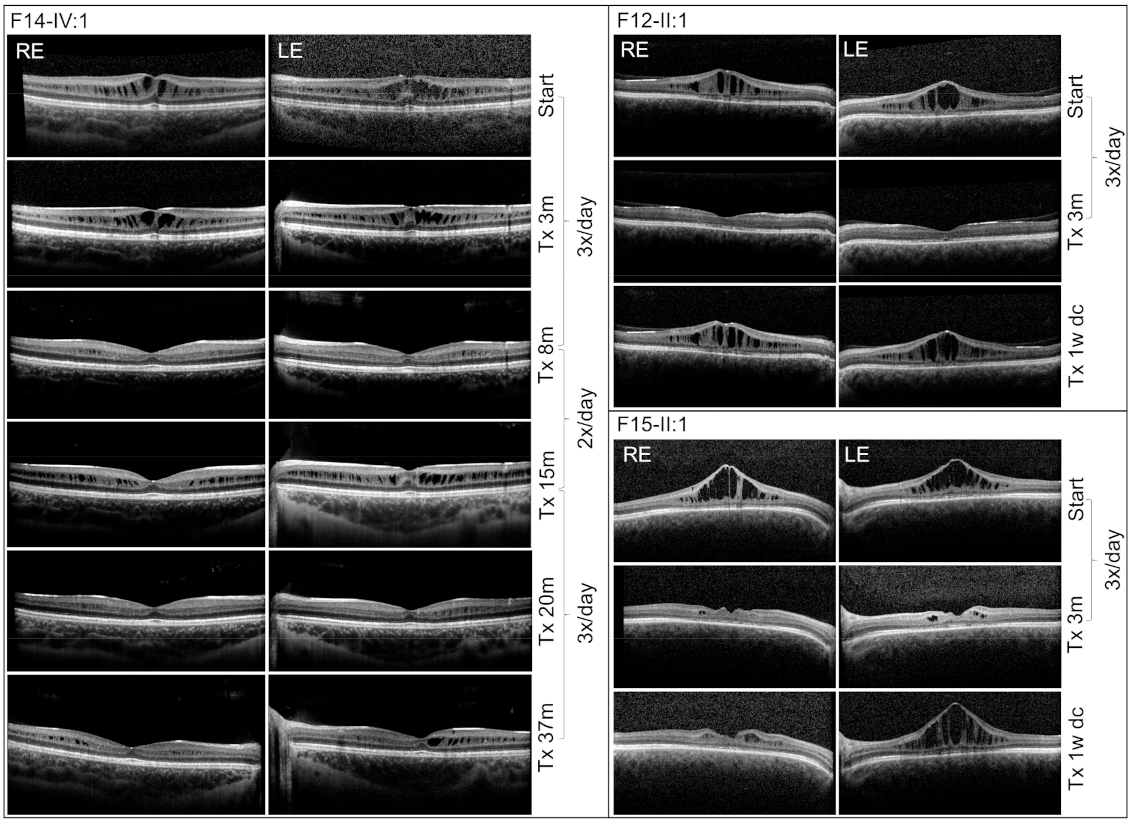
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molday, R.S.; Kellner, U.; Weber, B.H. X-linked juvenile retinoschisis: Clinical diagnosis, genetic analysis, and molecular mechanisms. Prog. Retin. Eye Res. 2012, 31, 195–212. [Google Scholar] [CrossRef] [PubMed]
- George, N.D.; Yates, J.R.; Moore, A.T. X linked retinoschisis. Br. J. Ophthalmol. 1995, 79, 697–702. [Google Scholar] [CrossRef]
- Hu, Q.R.; Huang, L.Z.; Chen, X.L.; Xia, H.K.; Li, T.Q.; Li, X.X. Genetic analysis and clinical features of X-linked retinoschisis in Chinese patients. Sci. Rep. 2017, 7, 44060. [Google Scholar] [CrossRef]
- Hinds, A.M.; Fahim, A.; Moore, A.T.; Wong, S.C.; Michaelides, M. Bullous X linked retinoschisis: Clinical features and prognosis. Br. J. Ophthalmol. 2018, 102, 622–624. [Google Scholar] [CrossRef] [PubMed]
- George, N.D.; Yates, J.R.; Moore, A.T. Clinical features in affected males with X-linked retinoschisis. Arch. Ophthalmol. 1996, 114, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Kellner, U.; Brümmer, S.; Foerster, M.H.; Wessing, A. X-linked congenital retinoschisis. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990, 228, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.K.; Liu, L.; Chen, H.M.; Tsai, S.; Chang, T.C.; Tsai, T.H.; Yang, C.M.; Chao, A.N.; Chen, K.J.; Kao, L.Y.; et al. Clinical presentations of X-linked retinoschisis in Taiwanese patients confirmed with genetic sequencing. Mol. Vis. 2015, 21, 487–501. [Google Scholar] [PubMed]
- Hotta, Y.; Nakamura, M.; Okamoto, Y.; Nomura, R.; Terasaki, H.; Miyake, Y. Different mutation of the XLRS1 gene causes juvenile retinoschisis with retinal white flecks. Br. J. Ophthalmol. 2001, 85, 238–239. [Google Scholar] [CrossRef] [PubMed]
- Fahim, A.T.; Ali, N.; Blachley, T.; Michaelides, M. Peripheral fundus findings in X-linked retinoschisis. Br. J. Ophthalmol. 2017, 101, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Apushkin, M.A.; Fishman, G.A.; Rajagopalan, A.S. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina 2005, 25, 612–618. [Google Scholar] [CrossRef]
- Pimenides, D.; George, N.D.; Yates, J.R.; Bradshaw, K.; Roberts, S.A.; Moore, A.T.; Trump, D. X-linked retinoschisis: Clinical phenotype and RS1 genotype in 86 UK patients. J. Med. Genet. 2005, 42, e35. [Google Scholar] [CrossRef] [PubMed]
- Khandhadia, S.; Trump, D.; Menon, G.; Lotery, A.J. X-linked retinoschisis maculopathy treated with topical dorzolamide, and relationship to genotype. Eye 2011, 25, 922–928. [Google Scholar] [CrossRef]
- Collison, F.T.; Genead, M.A.; Fishman, G.A.; Stone, E.M. Resolution of mid-peripheral schisis in x-linked retinoschisis with the use of dorzolamide. Ophthalmic Genet. 2014, 35, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Thobani, A.; Fishman, G.A. The use of carbonic anhydrase inhibitors in the retreatment of cystic macular lesions in retinitis pigmentosa and X-linked retinoschisis. Retina 2011, 31, 312–315. [Google Scholar] [CrossRef]
- Sauer, C.G.; Gehrig, A.; Warneke-Wittstock, R.; Marquardt, A.; Ewing, C.C.; Gibson, A.; Lorenz, B.; Jurklies, B.; Weber, B.H. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat. Genet. 1997, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, A.; Weber, B.H.; Lorenz, B.; Andrassi, M. First molecular evidence for a de novo mutation in RS1 (XLRS1) associated with X linked juvenile retinoschisis. J. Med. Genet. 1999, 36, 932–934. [Google Scholar] [PubMed]
- Khan, N.W.; Jamison, J.A.; Kemp, J.A.; Sieving, P.A. Analysis of photoreceptor function and inner retinal activity in juvenile X-linked retinoschisis. Vision Res. 2001, 41, 3931–3942. [Google Scholar] [CrossRef]
- Molday, L.L.; Hicks, D.; Sauer, C.G.; Weber, B.H.; Molday, R.S. Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Investig. Ophthalmol. Vis. Sci. 2001, 42, 816–825. [Google Scholar]
- Xiao, S.; Sun, W.; Xiao, X.; Li, S.; Luo, H.; Jia, X.; Ouyang, J.; Li, X.; Wang, Y.; Jiang, Y.; et al. Clinical and genetic features of retinoschisis in 120 families with RS1 mutations. Br. J. Ophthalmol. 2021. [Google Scholar] [CrossRef]
- Kim, D.Y.; Mukai, S. X-linked juvenile retinoschisis (XLRS): A review of genotype-phenotype relationships. Semin. Ophthalmol. 2013, 28, 392–396. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Li, S.; Jia, X.; Xiao, X.; Wang, P.; Guo, X.; Zhang, Q. Novel RS1 mutations associated with X-linked juvenile retinoschisis. Int. J. Mol. Med. 2012, 29, 644–648. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- The Retinoschisis Consortium. Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. Hum. Mol. Genet. 1998, 7, 1185–1192. [Google Scholar] [CrossRef]
- Huang, L.; Sun, L.; Wang, Z.; Chen, C.; Wang, P.; Sun, W.; Luo, X.; Ding, X. Clinical manifestation and genetic analysis in Chinese early onset X-linked retinoschisis. Mol. Genet. Genomic Med. 2020, 8, e1421. [Google Scholar] [CrossRef] [PubMed]
- Hotta, Y.; Fujiki, K.; Hayakawa, M.; Ohta, T.; Fujimaki, T.; Tamaki, K.; Yokoyama, T.; Kanai, A.; Hirakata, A.; Hida, T.; et al. Japanese juvenile retinoschisis is caused by mutations of the XLRS1 gene. Hum. Genet. 1998, 103, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Hiriyanna, K.T.; Bingham, E.L.; Yashar, B.M.; Ayyagari, R.; Fishman, G.; Small, K.W.; Weinberg, D.V.; Weleber, R.G.; Lewis, R.A.; Andreasson, S.; et al. Novel mutations in XLRS1 causing retinoschisis, including first evidence of putative leader sequence change. Hum. Mutat. 1999, 14, 423–427. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ko, H.S.; Yu, Y.S.; Hwang, J.M.; Lee, J.J.; Kim, S.Y.; Kim, J.Y.; Seong, M.W.; Park, K.H.; Park, S.S. Molecular genetic characteristics of X-linked retinoschisis in Koreans. Mol. Vis. 2009, 15, 833–843. [Google Scholar]
- Riveiro-Alvarez, R.; Trujillo-Tiebas, M.J.; Gimenez-Pardo, A.; Garcia-Hoyos, M.; Lopez-Martinez, M.A.; Aguirre-Lamban, J.; Garcia-Sandoval, B.; Vazquez-Fernandez del Pozo, S.; Cantalapiedra, D.; Avila-Fernandez, A.; et al. Correlation of genetic and clinical findings in Spanish patients with X-linked juvenile retinoschisis. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4342–4350. [Google Scholar] [CrossRef]
- Mashima, Y.; Shinoda, K.; Ishida, S.; Ozawa, Y.; Kudoh, J.; Iwata, T.; Oguchi, Y.; Shimizu, N. Identification of four novel mutations of the XLRS1 gene in Japanese patients with X-linked juvenile retinoschisis. Mutation in brief no. 234. Online. Hum. Mutat. 1999, 13, 338. [Google Scholar] [CrossRef]
- Inoue, Y.; Yamamoto, S.; Okada, M.; Tsujikawa, M.; Inoue, T.; Okada, A.A.; Kusaka, S.; Saito, Y.; Wakabayashi, K.; Miyake, Y.; et al. X-linked retinoschisis with point mutations in the XLRS1 gene. Arch. Ophthalmol. 2000, 118, 93–96. [Google Scholar] [CrossRef][Green Version]
- Curat, C.A.; Eck, M.; Dervillez, X.; Vogel, W.F. Mapping of epitopes in discoidin domain receptor 1 critical for collagen binding. J. Biol. Chem. 2001, 276, 45952–45958. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Waters, C.T.; Rothman, A.M.; Jakins, T.J.; Römisch, K.; Trump, D. Intracellular retention of mutant retinoschisin is the pathological mechanism underlying X-linked retinoschisis. Hum. Mol. Genet. 2002, 11, 3097–3105. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Mengher, L.S.; Tan, M.H.; Moore, A.T. An unusual fundus phenotype of inner retinal sheen in X-linked retinoschisis. Eye 2009, 23, 1876–1878. [Google Scholar] [CrossRef]
- Vijayasarathy, C.; Ziccardi, L.; Zeng, Y.; Smaoui, N.; Caruso, R.C.; Sieving, P.A. Null retinoschisin-protein expression from an RS1 c354del1-ins18 mutation causing progressive and severe XLRS in a cross-sectional family study. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5375–5383. [Google Scholar] [CrossRef]
- Kjellström, S.; Vijayasarathy, C.; Ponjavic, V.; Sieving, P.A.; Andréasson, S. Long-term 12 year follow-up of X-linked congenital retinoschisis. Ophthalmic. Genet. 2010, 31, 114–125. [Google Scholar] [CrossRef]
- Bennett, L.D.; Wang, Y.Z.; Klein, M.; Pennesi, M.E.; Jayasundera, T.; Birch, D.G. Structure/Psychophysical Relationships in X-Linked Retinoschisis. Investig. Ophthalmol. Vis. Sci. 2016, 57, 332–337. [Google Scholar] [CrossRef]
- Stringa, F.; Tsamis, E.; Papayannis, A.; Chwiejczak, K.; Jalil, A.; Biswas, S.; Ahmad, H.; Stanga, P.E. Segmented swept source optical coherence tomography angiography assessment of the perifoveal vasculature in patients with X-linked juvenile retinoschisis: A serial case report. Int. Med. Case. Rep. J. 2017, 10, 329–335. [Google Scholar] [CrossRef]
- Padrón-Pérez, N.; Català-Mora, J.; Díaz, J.; Arias, L.; Prat, J.; Caminal, J.M. Swept-source and optical coherence tomography angiography in patients with X-linked retinoschisis. Eye 2018, 32, 707–715. [Google Scholar] [CrossRef]
- Mastropasqua, R.; Toto, L.; Di Antonio, L.; Parodi, M.B.; Sorino, L.; Antonucci, I.; Stuppia, L.; Di Nicola, M.; Mariotti, C. Optical Coherence Tomography Angiography Findings in X-Linked Retinoschisis. Ophthalmic Surg. Lasers Imaging Retina 2018, 49, e20–e31. [Google Scholar] [CrossRef] [PubMed]
- Han, I.C.; Whitmore, S.S.; Critser, D.B.; Lee, S.Y.; DeLuca, A.P.; Daggett, H.T.; Affatigato, L.M.; Mullins, R.F.; Tucker, B.A.; Drack, A.V.; et al. Wide-Field Swept-Source OCT and Angiography in X-Linked Retinoschisis. Ophthalmol. Retina 2019, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Family | DNA Level | Protein Level | ClinVar Interpretation/VCV Accession | References |
|---|---|---|---|---|
| F1, F2 | c.20del | p.(Gly7Alafs*119) | Not present | Novel |
| F3, F4, F15 | c.33_36del | p.(Leu11Phefs*114) | Pathogenic/VCV000098944.7 | [25] |
| F5 | c.187T>C | p.(Cys63Arg) | Not present | [26] |
| F6 | c.275G>A | p.(Trp92*) | Not present | Novel |
| F7, F8 | c.305G>A | p.(Arg102Gln) | Pathogenic/VCV000009896.11 | [11,25,27,28,29] |
| F9 | c.375_379del | p.(Asp126Glufs*16) | Not present | Novel |
| F10 | c.539C>A | p.(Ser180*) | Not present | Novel |
| F16 | c.421C>T | p.(Arg141Cys) | Pathogenic/VCV000098959.9 | [11,25,30] |
| F11, F17 | c.544C>T | p.(Arg182Cys) | Pathogenic/VCV000098986.3 | [25,29,31,32] |
| F12 | c.574C>T | p.(Pro192Ser) | Pathogenic/VCV000098990.4 | [15,25,32] |
| F13 | c.575_576insT | p.(Ile194Hisfs*70) | Not present | Novel # |
| F14 | c.637C>T | p.(Arg213Trp) | Pathogenic/VCV000099009.5 | [25,29,33,34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kousal, B.; Hlavata, L.; Vlaskova, H.; Dvorakova, L.; Brichova, M.; Dubska, Z.; Langrova, H.; Vincent, A.L.; Dudakova, L.; Liskova, P. Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population. Genes 2021, 12, 1816. https://doi.org/10.3390/genes12111816
Kousal B, Hlavata L, Vlaskova H, Dvorakova L, Brichova M, Dubska Z, Langrova H, Vincent AL, Dudakova L, Liskova P. Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population. Genes. 2021; 12(11):1816. https://doi.org/10.3390/genes12111816
Chicago/Turabian StyleKousal, Bohdan, Lucia Hlavata, Hana Vlaskova, Lenka Dvorakova, Michaela Brichova, Zora Dubska, Hana Langrova, Andrea L. Vincent, Lubica Dudakova, and Petra Liskova. 2021. "Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population" Genes 12, no. 11: 1816. https://doi.org/10.3390/genes12111816
APA StyleKousal, B., Hlavata, L., Vlaskova, H., Dvorakova, L., Brichova, M., Dubska, Z., Langrova, H., Vincent, A. L., Dudakova, L., & Liskova, P. (2021). Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population. Genes, 12(11), 1816. https://doi.org/10.3390/genes12111816