
Prenatal Adversity Alters the Epigenetic Profile of the Prefrontal Cortex: Sexually Dimorphic Effects of Prenatal Alcohol Exposure and Food-Related Stress

 ,
,
Abstract
1. Introduction
2. Materials & Methods
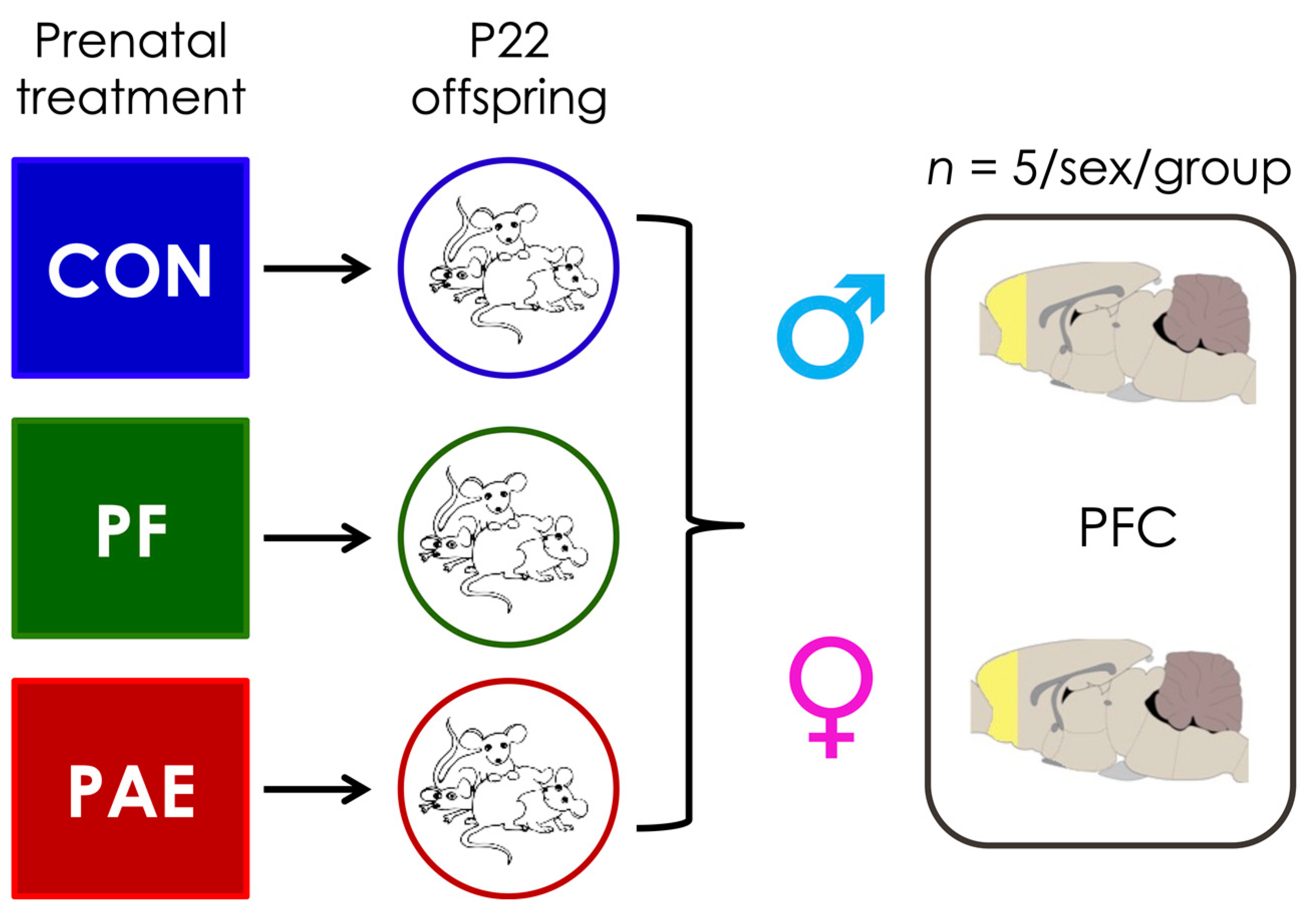
2.1. Prenatal Treatments
2.2. Sample Collection and DNA Extraction
2.3. Methylated DNA Immunoprecipitation and Next-Generation Sequencing
2.4. Bioinformatic Analyses
2.4.1. Next-Generation Sequencing Quality Control
2.4.2. Peakset Generation
2.4.3. Data Preprocessing and Normalization
2.5. Differentially Methylated Region (DMR) Identification
2.6. Genomic Enrichment
2.7. Gene Ontology Analyses
2.8. Chi-Squared Tests of the Direction of DMRs
3. Results
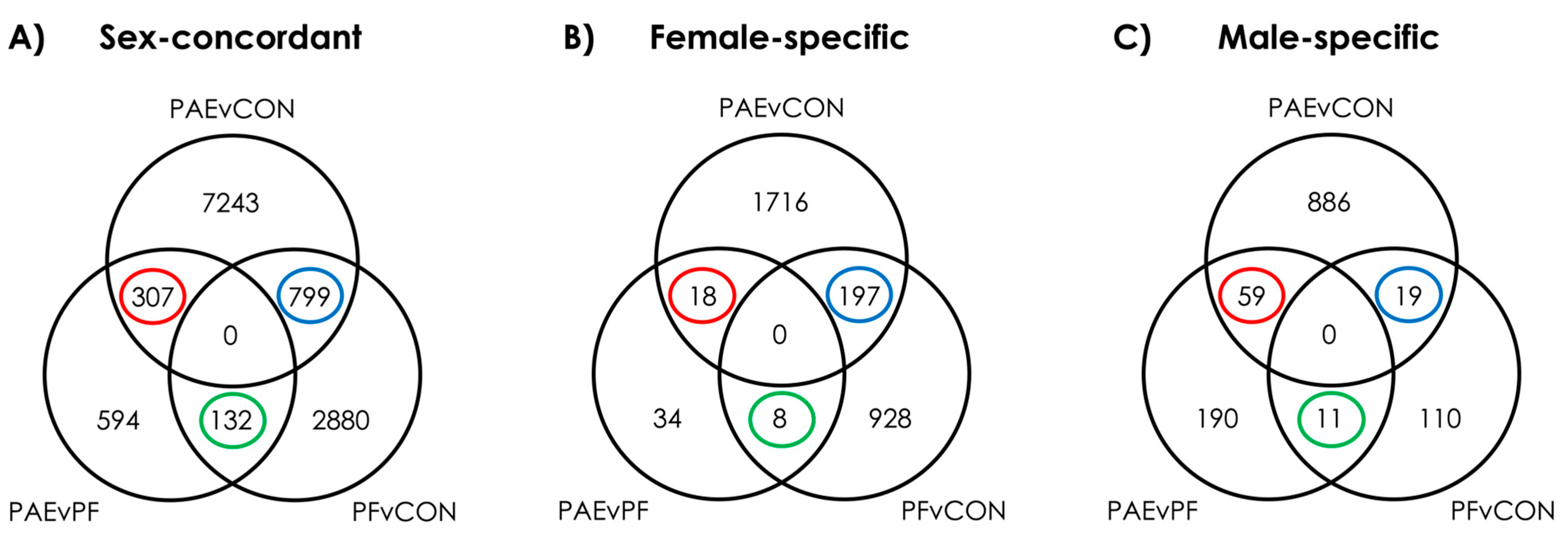
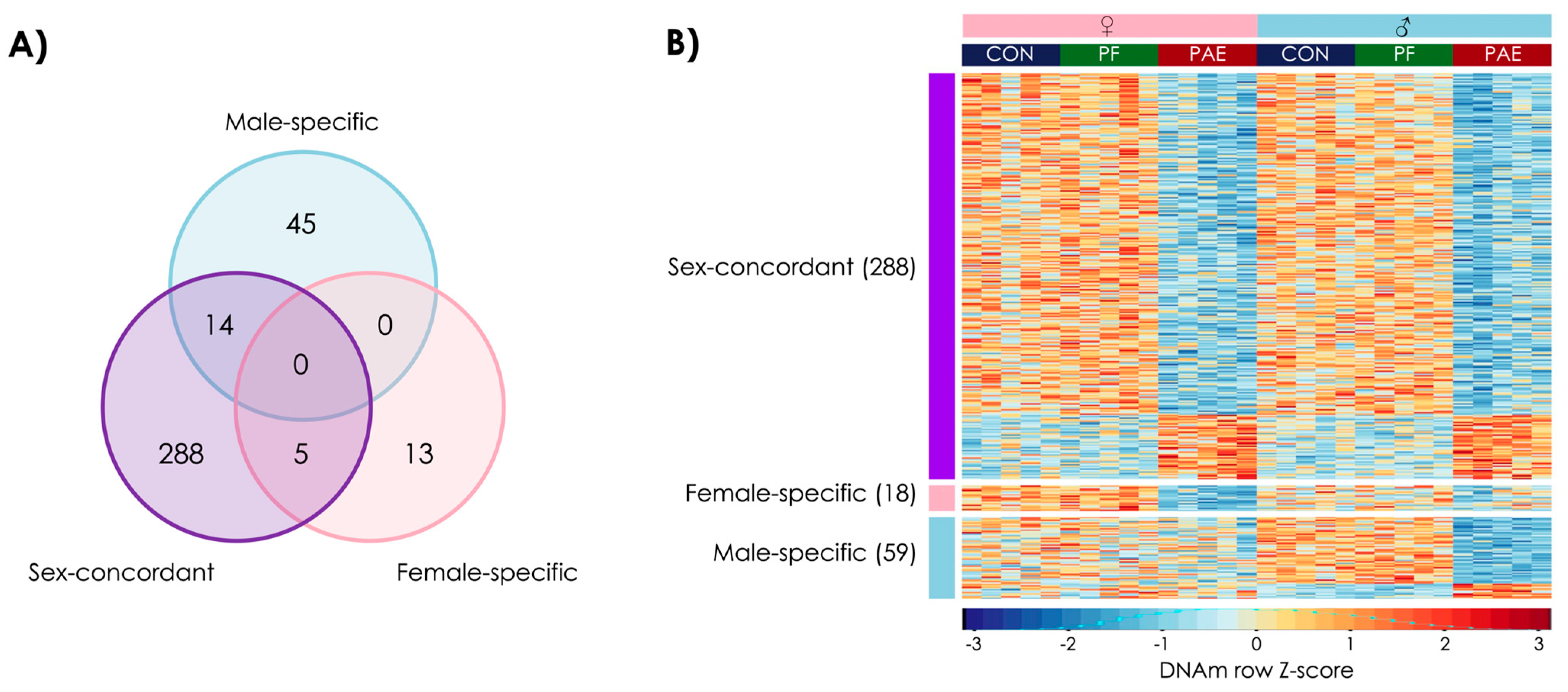
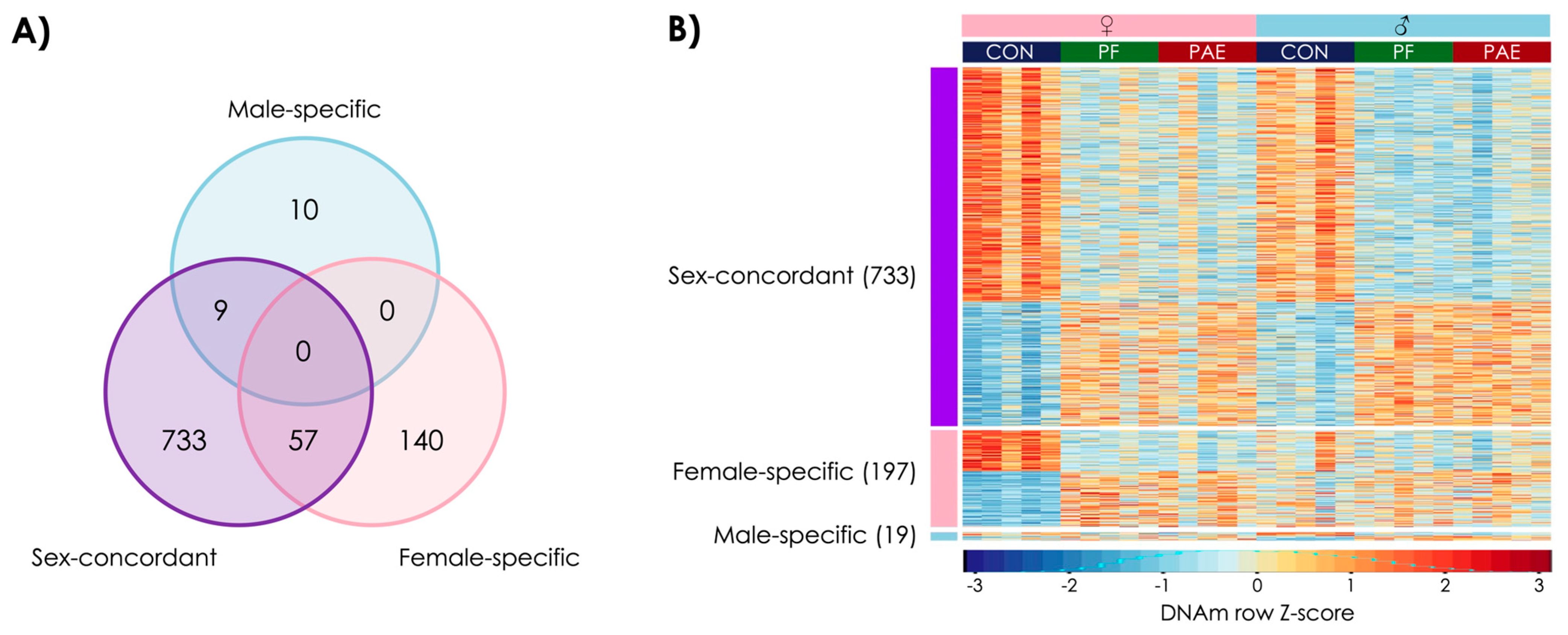
3.1. PAE Resulted in Sex-Concordant Alterations to DNAm Patterns
3.2. PAE Resulted in Sex-Specific Alterations to DNAm Patterns
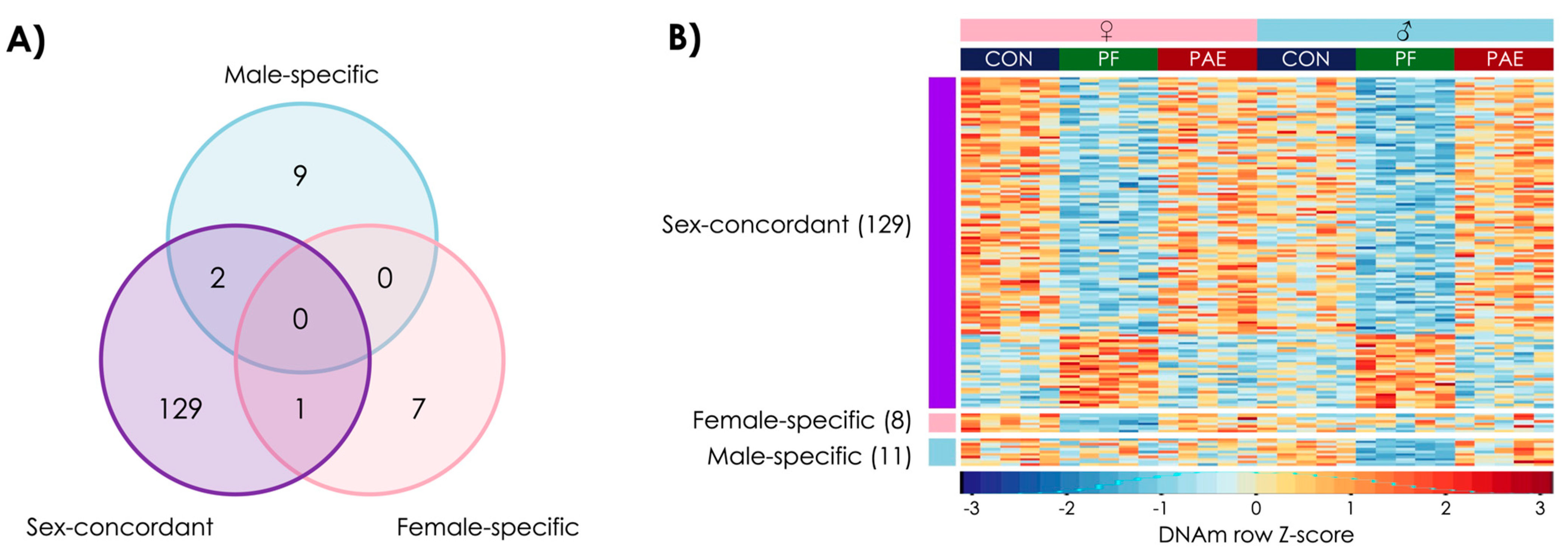
3.3. Prenatal Food-Related Stress Had Both Sex-Concordant and Sex-Specific Effects
3.4. PAE and PF Animals Shared Some Sex-Concordant and Sex-Specific Effects on DNAm Patterns
3.5. PAE-Specific and Shared DMRs Overlapped with Genes Linked to Autism Spectrum Disorder
4. Discussion
4.1. PAE-Specific Alterations
4.2. Prenatal Food-Related Stress-Induced Alterations
4.3. Common Impacts of Prenatal Stressors
4.4. Overlaps between Prenatal Stressors and Autism Spectrum Disorder
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ronald, A.; Pennell, C.E.; Whitehouse, A.J. Prenatal Maternal Stress Associated with ADHD and Autistic Traits in Early Childhood. Front. Psychol. 2011, 1, 223. [Google Scholar] [CrossRef] [PubMed]
- Beversdorf, D.Q.; Stevens, H.E.; Margolis, K.G.; Van de Water, J. Prenatal Stress and Maternal Immune Dysregulation in Autism Spectrum Disorders: Potential Points for Intervention. Curr. Pharm. Des. 2019, 25, 4331–4343. [Google Scholar] [CrossRef] [PubMed]
- Andalib, S.; Emamhadi, M.R.; Yousefzadeh-Chabok, S.; Shakouri, S.K.; Høilund-Carlsen, P.F.; Vafaee, M.S.; Michel, T.M. Maternal SSRI Exposure Increases the Risk of Autistic Offspring: A Meta-Analysis and Systematic Review. Eur. Psychiatry 2017, 45, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Karimi, P.; Kamali, E.; Mousavi, S.M.; Karahmadi, M. Environmental Factors Influencing the Risk of Autism. J. Res. Med. Sci. 2017, 22, 27. [Google Scholar]
- Bird, A. Perceptions of Epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Aristizabal, M.J.; Anreiter, I.; Halldorsdottir, T.; Odgers, C.L.; McDade, T.W.; Goldenberg, A.; Mostafavi, S.; Kobor, M.S.; Binder, E.B.; Sokolowski, M.B.; et al. Biological Embedding of Experience: A Primer on Epigenetics. Proc. Natl. Acad. Sci. USA 2020, 117, 23261. [Google Scholar] [CrossRef] [PubMed]
- Boyce, W.T.; Kobor, M.S. Development and the Epigenome: The ‘Synapse’ of Gene-Environment Interplay. Dev. Sci. 2015, 18, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Floyd, L.L.; Weber, M.K. Fetal Alcohol Syndrome Prevention Team, Division of Birth Defects and Developmental Disabilities, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention (CDC). Guidelines for identifying and referring persons with fetal alcohol syndrome. MMWR Recomm. Rep. 2005, 54, 1–4. [Google Scholar]
- Weinberg, J.; Sliwowska, J.H.; Lan, N.; Hellemans, K.G.C. Prenatal Alcohol Exposure: Foetal Programming, the Hypothalamic-Pituitary-Adrenal Axis and Sex Differences in Outcome. J. Neuroendocrinol. 2008, 20, 470–488. [Google Scholar] [CrossRef]
- Kaitlyn, M.; Rasmussen, C.; Oberlander, T.F.; Loock, C.; Pei, J.; Andrew, G.; Reynolds, J.; Weinberg, J. Dysregulation of the Cortisol Diurnal Rhythm Following Prenatal Alcohol Exposure and Early Life Adversity. Alcohol 2016, 53, 9–18. [Google Scholar]
- Kable, A.J.; Mehta, P.K.; Coles, C.D. Alterations in Insulin Levels in Adults with Prenatal Alcohol Exposure. Alcohol. Clin. Exp. Res. 2021, 45, 500–506. [Google Scholar]
- Hellemans, K.G.; Sliwowska, J.H.; Verma, P.; Weinberg, J. Prenatal Alcohol Exposure: Fetal Programming and Later Life Vulnerability to Stress, Depression and Anxiety Disorders. Neurosci. Biobehav. Rev. 2010, 34, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, T.S.; Weinberg, J. Prenatal Alcohol Exposure: Impact on Neuroendocrine—Neuroimmune Networks; Springer: Boston, MA, USA, 2013; pp. 307–357. [Google Scholar]
- Glover, V. Maternal Depression, Anxiety and Stress During Pregnancy and Child Outcome; What Needs to Be Done. Best Pract. Res. Clin. Obstet. Gynaecol. 2014, 28, 25–35. [Google Scholar] [CrossRef]
- Van den Bergh, B.R.H.; van den Heuvel, M.I.; Lahti, M.; Braeken, M.; de Rooij, S.R.; Entringer, S.; Hoyer, D.; Roseboom, T.; Räikkönen, K.; King, S.; et al. Prenatal Developmental Origins of Behavior and Mental Health: The Influence of Maternal Stress in Pregnancy. Neurosci. Biobehav. Rev. 2020, 117, 26–64. [Google Scholar] [CrossRef]
- Ruggiero, M.J.; Boschen, K.E.; Roth, T.L.; Klintsova, A.Y. Sex Differences in Early Postnatal Microglial Colonization of the Developing Rat Hippocampus Following a Single-Day Alcohol Exposure. J. Neuroimmune Pharmacol. 2018, 13, 189–203. [Google Scholar] [CrossRef]
- Uban, K.A.; Comeau, W.L.; Ellis, L.A.; Galea, L.A.; Weinberg, J. Basal Regulation of HPA and Dopamine Systems Is Altered Differentially in Males and Females by Prenatal Alcohol Exposure and Chronic Variable Stress. Psychoneuroendocrinology 2013, 38, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rivier, C. Gender Differences in the Effect of Prenatal Alcohol Exposure on the Hypothalamic-Pituitary-Adrenal Axis Response to Immune Signals. Psychoneuroendocrinology 1996, 21, 145–155. [Google Scholar] [CrossRef]
- Kelly, S.J.; Day, N.; Streissguth, A.P. Effects of Prenatal Alcohol Exposure on Social Behavior in Humans and Other Species. Neurotoxicology Teratol. 2000, 22, 143–149. [Google Scholar] [CrossRef]
- Holman, P.J.; Baglot, S.L.; Morgan, E.; Weinberg, J. Effects of Prenatal Alcohol Exposure on Social Competence: Asymmetry in Play Partner Preference among Heterogeneous Triads of Male and Female Rats. Dev. Psychobiol. 2019, 61, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Holman, P.J.; Raineki, C.; Chao, A.; Grewal, R.; Haghighat, S.; Fung, C.; Morgan, E.; Ellis, L.; Yu, W.; Weinberg, J. Altered Social Recognition Memory and Hypothalamic Neuropeptide Expression in Adolescent Male and Female Rats Following Prenatal Alcohol Exposure and/or Early-Life Adversity. Psychoneuroendocrinology 2021, 126, 105146. [Google Scholar] [CrossRef]
- Caldwell, K.K.; Sheema, S.; Paz, R.D.; Samudio-Ruiz, S.L.; Laughlin, M.H.; Spence, N.E.; Roehlk, M.J.; Alcon, S.N.; Allan, A.M. Fetal Alcohol Spectrum Disorder-Associated Depression: Evidence for Reductions in the Levels of Brain-Derived Neurotrophic Factor in a Mouse Model. Pharmacol. Biochem. Behav. 2008, 90, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Raineki, C.; Hellemans, K.G.C.; Bodnar, T.; Lavigne, K.M.; Ellis, L.; Woodward, T.S.; Weinberg, J. Neurocircuitry Underlying Stress and Emotional Regulation in Animals Prenatally Exposed to Alcohol and Subjected to Chronic Mild Stress in Adulthood. Front. Endocrinol. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.Y.Y.; Raineki, C.; Ellis, L.; Yu, W.; Weinberg, J. Interactive Effects of Prenatal Alcohol Exposure and Chronic Stress in Adulthood on Anxiety-Like Behavior and Central Stress-Related Receptor mRNA Expression: Sex- and Time-Dependent Effects. Psychoneuroendocrinology 2018, 97, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.X.; Jonsson, E.; Salmon, A.; Sebastianski, M. Incidence and Prevalence of Fetal Alcohol Spectrum Disorder by Sex and Age Group in Alberta, Canada. J. Popul. Ther. Clin. Pharmacol. 2014, 21, e395–e404. [Google Scholar] [PubMed]
- Woods, K.J.; Thomas, K.G.F.; Molteno, C.D.; Jacobson, J.L.; Jacobson, S.W.; Meintjes, E.M. Prenatal Alcohol Exposure Affects Brain Function During Place Learning in a Virtual Environment Differently in Boys and Girls. Brain Behav. 2018, 8, e01103. [Google Scholar] [CrossRef] [PubMed]
- Tesche, C.D.; Kodituwakku, P.W.; Garcia, C.M.; Houck, J.M. Sex-Related Differences in Auditory Processing in Adolescents with Fetal Alcohol Spectrum Disorder: A Magnetoencephalographic Study. NeuroImage 2015, 7, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Uban, K.A.; Herting, M.M.; Wozniak, J.R.; Sowell, E.R. Sex Differences in Associations between White Matter Microstructure and Gonadal Hormones in Children and Adolescents with Prenatal Alcohol Exposure. Psychoneuroendocrinology 2017, 83, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Sayal, K.; Heron, J.; Golding, J.; Emond, A. Prenatal Alcohol Exposure and Gender Differences in Childhood Mental Health Problems: A Longitudinal Population-Based Study. Pediatrics 2007, 119, e426–e434. [Google Scholar] [CrossRef]
- Lussier, A.A.; Weinberg, J.; Kobor, M.S. Epigenetics Studies of Fetal Alcohol Spectrum Disorder: Where Are We Now? Epigenomics 2017, 9, 291–311. [Google Scholar] [CrossRef]
- Petrelli, B.; Weinberg, J.; Hicks, G.G. Effects of Prenatal Alcohol Exposure (PAE): Insights into FASD Using Mouse Models of PAE. Biochem. Cell Biol. 2018, 96, 131–147. [Google Scholar] [CrossRef]
- Schaffner, S.L.; Lussier, A.A.; Baker, J.A.; Goldowitz, D.; Hamre, K.M.; Kobor, M.S. Neonatal Alcohol Exposure in Mice Induces Select Differentiation- and Apoptosis-Related Chromatin Changes Both Independent of and Dependent on Sex. Front. Genet. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Ngai, Y.F.; Sulistyoningrum, D.C.; O’Neill, R.; Innis, S.M.; Weinberg, J.; Devlin, A.M. Prenatal Alcohol Exposure Alters Methyl Metabolism and Programs Serotonin Transporter and Glucocorticoid Receptor Expression in Brain. Am. J. Physiology Regul. Integr. Comp. Physiol. 2015, 309, R613–R622. [Google Scholar] [CrossRef] [PubMed]
- Gatev, E.; Inkster, A.M.; Negri, G.L.; Konwar, C.; Lussier, A.A.; Skakkebaek, A.; Sokolowski, M.B.; Gravholt, C.H.; Dunn, E.C.; Kobor, M.S.; et al. Autosomal Sex-Associated Co-Methylated Regions Predict Biological Sex from DNA Methylation. Nucleic Acids Res. 2021, 49, 9097–9116. [Google Scholar] [PubMed]
- Glavas, M.M.; Ellis, L.; Yu, W.K.; Weinberg, J. Effects of Prenatal Ethanol Exposure on Basal Limbic-Hypothalamic-Pituitary-Adrenal Regulation: Role of Corticosterone. Alcohol. Clin. Exp. Res. 2007, 31, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Lan, N.; Chiu, M.P.Y.; Ellis, L.; Weinberg, J. Prenatal Alcohol Exposure and Prenatal Stress Differentially Alter Glucocorticoid Signaling in the Placenta and Fetal Brain. Neuroscience 2017, 342, 167–179. [Google Scholar] [CrossRef]
- Sliwowska, J.H.; Comeau, W.L.; Bodnar, T.S.; Ellis, L.; Weinberg, J. Prenatal Alcohol Exposure and Pair Feeding Differentially Impact Puberty and Reproductive Development in Female Rats: Role of the Kisspeptin System. Alcohol. Clin. Exp. Res. 2016, 40, 2368–2376. [Google Scholar] [CrossRef][Green Version]
- Gawałek, M.; Sliwowska, J.H. Neuronal Basis of Reproductive Dysfunctions Associated with Diet and Alcohol: From the Womb to Adulthood. Reprod. Biol. 2015, 15, 69–78. [Google Scholar] [CrossRef]
- Bodnar, T.S.; Hill, L.A.; Weinberg, J. Evidence for an Immune Signature of Prenatal Alcohol Exposure in Female Rats. Brain Behav. Immun. 2016, 58, 130–141. [Google Scholar] [CrossRef]
- Lumey, L.H.; Stein, A.D.; Kahn, H.S.; Romijn, J.A. Lipid Profiles in Middle-Aged Men and Women after Famine Exposure During Gestation: The Dutch Hunger Winter Families Study. Am. J. Clin. Nutr. 2009, 89, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Dearden, L.; Bouret, S.G.; Susan, E. Ozanne. Sex and Gender Differences in Developmental Programming of Metabolism. Mol. Metab. 2018, 15, 8–19. [Google Scholar]
- de Rooij, S.R.; Caan, M.W.A.; Swaab, D.F.; Nederveen, A.J.; Majoie, C.B.; Schwab, M.; Painter, R.C.; Roseboom, T.J. Prenatal Famine Exposure Has Sex-Specific Effects on Brain Size. Brain 2016, 139, 2136–2142. [Google Scholar] [CrossRef]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA Methylation Signatures Link Prenatal Famine Exposure to Growth and Metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent Epigenetic Differences Associated with Prenatal Exposure to Famine in Humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA Methylation Differences after Exposure to Prenatal Famine are Common and Timing- and Sex-Specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.M.; Gratton, A. Prefrontal Cortical Regulation of Hypothalamic-Pituitary-Adrenal Function in the Rat and Implications for Psychopathology: Side Matters. Psychoneuroendocrinology 2002, 27, 99–114. [Google Scholar] [CrossRef]
- Damasio, A.R.; Eighth, C.U. Ariens Kappers Lecture. The fabric of the mind: A neurobiological perspective. Prog. Brain Res. 2000, 126, 457–467. [Google Scholar] [PubMed]
- McKlveen, J.M.; Myers, B.; Flak, J.N.; Bundzikova, J.; Solomon, M.B.; Seroogy, K.B.; Herman, J.P. Role of Prefrontal Cortex Glucocorticoid Receptors in Stress and Emotion. Biol. Psychiatry 2013, 74, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, J.J.; Almeida, O.F.; Sousa, N. The stressed prefrontal cortex. Left? Right! Brain Behav. Immun. 2008, 22, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Bishop, S.; Gahagan, S.; Lord, C. Re-Examining the Core Features of Autism: A Comparison of Autism Spectrum Disorder and Fetal Alcohol Spectrum Disorder. J. Child Psychol. Psychiatry Allied Discip. 2007, 48, 1111–1121. [Google Scholar] [CrossRef]
- Popova, S.; Lange, S.; Shield, K.; Mihic, A.; Chudley, A.E.; Mukherjee, R.A.S.; Bekmuradov, D.; Rehm, J. Comorbidity of Fetal Alcohol Spectrum Disorder: A Systematic Review and Meta-Analysis. Lancet 2016, 387, 978–987. [Google Scholar] [CrossRef]
- Lange, S.; Rehm, J.; Anagnostou, E.; Popova, S. Prevalence of Externalizing Disorders and Autism Spectrum Disorders among Children with Fetal Alcohol Spectrum Disorder: Systematic Review and Meta-Analysis. Biochem. Cell Biol. 2018, 96, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Lussier, A.A.; Bodnar, T.S.; Mingay, M.; Morin, A.M.; Hirst, M.; Kobor, M.S.; Weinberg, J. Prenatal Alcohol Exposure: Profiling Developmental DNA Methylation Patterns in Central and Peripheral Tissues. Front. Genet. 2018, 9, 610. [Google Scholar] [CrossRef]
- Taiwo, O.; Wilson, G.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L. Methylome Analysis Using MeDIP-Seq with Low DNA Concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Stark, R.; Brown, G. Diffbind: Differential Binding Analysis of ChIP-Seq Peak Data. Bioconductor. 2011. Available online: https://bioconductor.org/packages/release/bioc/html/DiffBind.html (accessed on 8 October 2021).
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential Oestrogen Receptor Binding Is Associated with Clinical Outcome in Breast Cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gillis, J.; Mistry, M.; Pavlidis, P. Gene function analysis in complex data sets using ErmineJ. In Nature Protocols; Department of Psychiatry, University of British Columbia: Vancouver, BC, Canada, 2010; pp. 1148–1159. [Google Scholar]
- Lee, H.K.; Braynen, W.; Keshav, K.; Pavlidis, P. ErmineJ: Tool for Functional Analysis of Gene Expression Data Sets. BMC Bioinform. 2005, 6, 269. [Google Scholar] [CrossRef] [PubMed]
- Lussier, A.A.; Morin, A.M.; MacIsaac, J.L.; Salmon, J.; Weinberg, J.; Reynolds, J.N.; Pavlidis, P.; Chudley, A.E.; Kobor, M.S. DNA Methylation as a Predictor of Fetal Alcohol Spectrum Disorder. Clin. Epigenetics 2018, 10, 5. [Google Scholar] [CrossRef]
- Portales-Casamar, E.; Lussier, A.A.; Jones, M.J.; MacIsaac, J.L.; Edgar, R.D.; Mah, S.M.; Barhdadi, A.; Provost, S.; Lemieux-Perreault, L.; Cynader, M.S.; et al. DNA Methylation Signature of Human Fetal Alcohol Spectrum Disorder. Epigenetics Chromatin 2016, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Fransquet, P.D.; Hutchinson, D.; Olsson, C.A.; Wilson, J.; Allsop, S.; Najman, J.; Elliott, E.; Mattick, R.P.; Saffery, R.; Ryan, J. Perinatal Maternal Alcohol Consumption and Methylation of the Dopamine Receptor DRD4 in the Offspring: The Triple B Study. Environ. Epigenetics 2016, 2, dvw023. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of Common Genetic Risk Variants for Autism Spectrum Disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef]
- Hannon, E.; Schendel, D.; Ladd-Acosta, C.; Grove, J.; Agerbo, E.; Als, T.D.; Belliveau, R.; Bybjerg-Grauholm, J.; Bækved-Hansen, M.; Børglum, A.; et al. Elevated Polygenic Burden for Autism Is Associated with Differential DNA Methylation at Birth. Genome Med. 2018, 10, 19. [Google Scholar] [CrossRef]
- Andrews, S.V.; Sheppard, B.; Windham, G.C.; Schieve, L.A.; Schendel, D.E.; Croen, L.A.; Chopra, P.; Alisch, R.S.; Newschaffer, C.J.; Warren, S.T.; et al. Case-Control Meta-Analysis of Blood DNA Methylation and Autism Spectrum Disorder. Mol. Autism 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Berko, E.R.; Suzuki, M.; Beren, F.; Lemetre, C.; Alaimo, C.M.; Calder, R.B.; Ballaban-Gil, K.; Gounder, B.; Kampf, K.; Kirschen, J.; et al. Mosaic Epigenetic Dysregulation of Ectodermal Cells in Autism Spectrum Disorder. PLoS Genet. 2014, 10, e1004402. [Google Scholar] [CrossRef] [PubMed]
- Nardone, S.; Sams, D.S.; Zito, A.; Reuveni, E.; Elliott, E. Dysregulation of Cortical Neuron DNA Methylation Profile in Autism Spectrum Disorder. Cereb. Cortex 2017, 27, 5739–5754. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W.; et al. Genome-Wide DNA Methylation Profiling Identifies Convergent Molecular Signatures Associated with Idiopathic and Syndromic Autism in Post-Mortem Human Brain Tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA Methylation Alterations in Multiple Brain Regions in Autism. Mol. Psychiatry 2014, 19, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Otero, N.K.H.; Thomas, J.D.; Saski, C.A.; Xia, X.; Kelly, S.J. Choline Supplementation and DNA Methylation in the Hippocampus and Prefrontal Cortex of Rats Exposed to Alcohol during Development. Alcohol. Clin. Exp. Res. 2012, 36, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, J.R.; Fink, B.A.; Fuglestad, A.J.; Eckerle, J.K.; Boys, C.J.; Sandness, K.E.; Radke, J.P.; Miller, N.C.; Lindgren, C.; Brearley, A.M.; et al. Four-Year Follow-up of a Randomized Controlled Trial of Choline for Neurodevelopment in Fetal Alcohol Spectrum Disorder. J. Neurodev. Disord. 2020, 12, 9. [Google Scholar] [CrossRef]
- Wozniak, J.R.; Fuglestad, A.J.; Eckerle, J.K.; Fink, B.A.; Hoecker, H.L.; Boys, C.J.; Radke, J.P.; Kroupina, M.G.; Miller, N.C.; Brearley, A.M.; et al. Choline Supplementation in Children with Fetal Alcohol Spectrum Disorders: A Randomized, Double-Blind, Placebo-Controlled Trial. Am. J. Clin. Nutr. 2015, 102, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, D.; Jha, S.K.; Jha, N.K.; Ambasta, R.K. Chapter three—Ion channels in neurological disorders. Adv. Protein Chem. Struct. Biol. 2016, 103, 97–136. [Google Scholar] [PubMed]
- Noh, W.; Pak, S.; Choi, G.; Yang, S.; Yang, S. Transient Potassium Channels: Therapeutic Targets for Brain Disorders. Front. Cell. Neurosci. 2019, 13, 265. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Takeichi, M. Cadherins in Brain Morphogenesis and Wiring. Physiol. Rev. 2012, 92, 597–634. [Google Scholar] [CrossRef]
- Schmid, R.S.; Anton, E.S. Role of Integrins in the Development of the Cerebral Cortex. Cereb. Cortex 2003, 13, 219–224. [Google Scholar] [CrossRef]
- Mukhtar, T.; Breda, J.; Grison, A.; Karimaddini, Z.; Grobecker, P.; Iber, D.; Beisel, C.; van Nimwegen, E.; Taylor, V. Tead Transcription Factors Differentially Regulate Cortical Development. Sci. Rep. 2020, 10, 4625. [Google Scholar] [CrossRef]
- Wang, H.; Liu, F.; Chen, W.; Sun, X.; Cui, W.; Dong, Z.; Zhao, K.; Zhang, H.; Li, H.; Xing, G.; et al. Genetic Recovery of ErbB4 in Adulthood Partially Restores Brain Functions in Null Mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13105. [Google Scholar] [CrossRef]
- Lebel, C.; Mattson, S.N.; Riley, E.P.; Jones, K.L.; Adnams, C.M.; May, P.A.; Bookheimer, S.Y.; O’Connor, M.J.; Narr, K.L.; Kan, E.; et al. A Longitudinal Study of the Long-Term Consequences of Drinking During Pregnancy: Heavy in Utero Alcohol Exposure Disrupts the Normal Processes of Brain Development. J. Neurosci. 2012, 32, 15243–15251. [Google Scholar] [CrossRef]
- Treit, S.; Chen, Z.; Zhou, D.; Baugh, L.; Rasmussen, C.; Andrew, G.; Pei, J.; Beaulieu, C. Sexual Dimorphism of Volume Reduction but Not Cognitive Deficit in Fetal Alcohol Spectrum Disorders: A Combined Diffusion Tensor Imaging, Cortical Thickness and Brain Volume Study. NeuroImage 2017, 15, 284–297. [Google Scholar] [CrossRef]
- Gambin, T.; Yuan, B.; Bi, W.; Liu, P.; Rosenfeld, J.A.; Coban-Akdemir, Z.; Pursley, A.N.; Nagamani, S.C.S.; Marom, R.; Golla, S.; et al. Identification of Novel Candidate Disease Genes from De Novo Exonic Copy Number Variants. Genome Med. 2017, 9, 83. [Google Scholar] [CrossRef]
- Weinberg, J. Nutritional Issues in Perinatal Alcohol Exposure. Neurobehav. Toxicol. Teratol. 1984, 6, 261–269. [Google Scholar]
- Burdge, G.C.; Slater-Jefferies, J.; Torrens, C.; Phillips, E.S.; Hanson, M.A.; Lillycrop, K.A. Dietary Protein Restriction of Pregnant Rats in the F0 Generation Induces Altered Methylation of Hepatic Gene Promoters in the Adult Male Offspring in the F1 and F2 Generations. Br. J. Nutr. 2007, 97, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Susser, E.S.; Lin, S.P.; Neugebauer, R.; Gorman, J.M. Increased Risk of Affective Disorders in Males after Second Trimester Prenatal Exposure to the Dutch Hunger Winter of 1944–45. Br. J. Psychiatry 1995, 166, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J. Effects of Ethanol and Maternal Nutritional Status on Fetal Development. Alcohol. Clin. Exp. Res. 1985, 9, 49–55. [Google Scholar] [CrossRef]
- Bodnar, T.S.; Raineki, C.; Wertelecki, W.; Yevtushok, L.; Plotka, L.; Zymak-Zakutnya, N.; Honerkamp-Smith, G.; Wells, A.; Rolland, M.; Woodward, T.S.; et al. Altered Maternal Immune Networks Are Associated with Adverse Child Neurodevelopment: Impact of Alcohol Consumption During Pregnancy. Brain Behav. Immun. 2018, 73, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, M.T.; Monk, C. Impact of Maternal Stress, Depression and Anxiety on Fetal Neurobehavioral Development. Clin. Obstet. Gynecol. 2009, 52, 425–440. [Google Scholar] [CrossRef]
- Cao-Lei, L.; Laplante, D.P.; King, S. Prenatal Maternal Stress and Epigenetics: Review of the Human Research. Curr. Mol. Biol. Rep. 2016, 2, 16–25. [Google Scholar] [CrossRef]
- Moon, A.L.; Haan, N.; Wilkinson, L.S.; Thomas, K.L.; Hall, J. CACNA1C: Association with Psychiatric Disorders, Behavior, and Neurogenesis. Schizophr. Bull. 2018, 44, 958–965. [Google Scholar] [CrossRef]
- Dedic, N.; Pöhlmann, M.L.; Richter, J.S.; Mehta, D.; Czamara, D.; Metzger, M.W.; Dine, J.; Bedenk, B.T.; Hartmann, J.; Wagner, K.V.; et al. Cross-Disorder Risk Gene CACNA1C Differentially Modulates Susceptibility to Psychiatric Disorders During Development and Adulthood. Mol. Psychiatry 2018, 23, 533–543. [Google Scholar] [CrossRef]
- Xiao, X.; Zheng, F.; Chang, H.; Ma, Y.; Yao, Y.; Luo, X.; Li, M. The Gene Encoding Protocadherin 9 (PCDH9), a Novel Risk Factor for Major Depressive Disorder. Neuropsychopharmacology 2018, 43, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Stevens, S.A.; Nash, K.; Koren, G.; Rovet, J. Autism Characteristics in Children with Fetal Alcohol Spectrum Disorders. Child Neuropsychol. 2013, 19, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.R.; MacKay, L.L.; Osborn, J.A. Autistic Behaviors in Offspring of Mothers Abusing Alcohol and Other Drugs: A Series of Case Reports. Alcohol. Clin. Exp. Res. 1995, 19, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Nanson, J.L. Autism in Fetal Alcohol Syndrome: A Report of Six Cases. Alcohol. Clin. Exp. Res. 1992, 16, 558–565. [Google Scholar] [CrossRef]
- Mukherjee, R.; Layton, M.; Yacoub, E.; Turk, J. Autism and Autistic Traits in People Exposed to Heavy Prenatal Alcohol: Data from a Clinical Series of 21 Individuals and Nested Case Control Study. Adv. Ment. Health Intellect. Disabil. 2011, 5, 42–49. [Google Scholar] [CrossRef]
- Singh, K.; Jayaram, M.; Kaare, M.; Leidmaa, E.; Jagomäe, T.; Heinla, I.; Hickey, M.A.; Kaasik, A.; Schäfer, M.K.; Innos, J.; et al. Neural Cell Adhesion Molecule Negr1 Deficiency in Mouse Results in Structural Brain Endophenotypes and Behavioral Deviations Related to Psychiatric Disorders. Sci. Rep. 2019, 9, 5457. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Greenberg, M.V.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Sample | Age at Collection | Tissue | Overlapping Genes | ||
|---|---|---|---|---|---|---|
| PAE | PF | Shared | ||||
| Genome-wide association study (GWAS) | ||||||
| Grove 2019 [65] | 18,381 ASD cases 27,969 controls | NEGR1* | MMS22L | |||
| Epigenome-wide association studies (EWAS) | ||||||
| Andrews 2018 [67] | 796 ASD cases 858 controls | 4–18 years | Blood | |||
| Berko 2014 [68] | 47 ASD cases 48 controls | 2–17 years | Buccal epithelial cells | NRG2+ | ||
| Hannon 2018 [66] | 629 ASD cases 634 controls | Birth | Neonatal blood spots | |||
| Ladd Acosta 2014 [71] | 19 ASD cases 21 controls | 2–51 years | Prefrontal cortex; Temporal cortex; Cerebellum | |||
| Nardone 2017 [69] | 16 male ASD cases 15 male controls | 17–68 years | Frontal cortex | NEDD4L | ||
| Wong 2019 [70] | 36 ASD cases 33 controls | 29.3 years (±28.2) | Prefrontal cortex | CDH13+ | PRKAR1B | |
| 33 ASD cases 38 controls | 29.1 years (±18.6) | Temporal cortex | ||||
| 34 ASD cases 29 controls | 30.6 years (±21.2) | Cerebellum | ||||
| 30 ASD cases 29 controls | 29.0 years (±18.9) | Prefrontal and temporal cortex, analyzed together | GRIK1 | FRMD4A | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lussier, A.A.; Bodnar, T.S.; Moksa, M.; Hirst, M.; Kobor, M.S.; Weinberg, J. Prenatal Adversity Alters the Epigenetic Profile of the Prefrontal Cortex: Sexually Dimorphic Effects of Prenatal Alcohol Exposure and Food-Related Stress. Genes 2021, 12, 1773. https://doi.org/10.3390/genes12111773
Lussier AA, Bodnar TS, Moksa M, Hirst M, Kobor MS, Weinberg J. Prenatal Adversity Alters the Epigenetic Profile of the Prefrontal Cortex: Sexually Dimorphic Effects of Prenatal Alcohol Exposure and Food-Related Stress. Genes. 2021; 12(11):1773. https://doi.org/10.3390/genes12111773
Chicago/Turabian StyleLussier, Alexandre A., Tamara S. Bodnar, Michelle Moksa, Martin Hirst, Michael S. Kobor, and Joanne Weinberg. 2021. "Prenatal Adversity Alters the Epigenetic Profile of the Prefrontal Cortex: Sexually Dimorphic Effects of Prenatal Alcohol Exposure and Food-Related Stress" Genes 12, no. 11: 1773. https://doi.org/10.3390/genes12111773
APA StyleLussier, A. A., Bodnar, T. S., Moksa, M., Hirst, M., Kobor, M. S., & Weinberg, J. (2021). Prenatal Adversity Alters the Epigenetic Profile of the Prefrontal Cortex: Sexually Dimorphic Effects of Prenatal Alcohol Exposure and Food-Related Stress. Genes, 12(11), 1773. https://doi.org/10.3390/genes12111773