
Alternative Lengthening of Telomeres: Lessons to Be Learned from Telomeric DNA Double-Strand Break Repair


{kind=link}
Abstract
:1. Aberrant Telomeric BIR—A Potential Route to Cancer
2. The Alternative Lengthening of Telomeres Pathway
3. BIR at Telomeres Underpins the ALT Pathway
4. ALT-Associated Nuclear Bodies—Sites of Telomere Recombination?
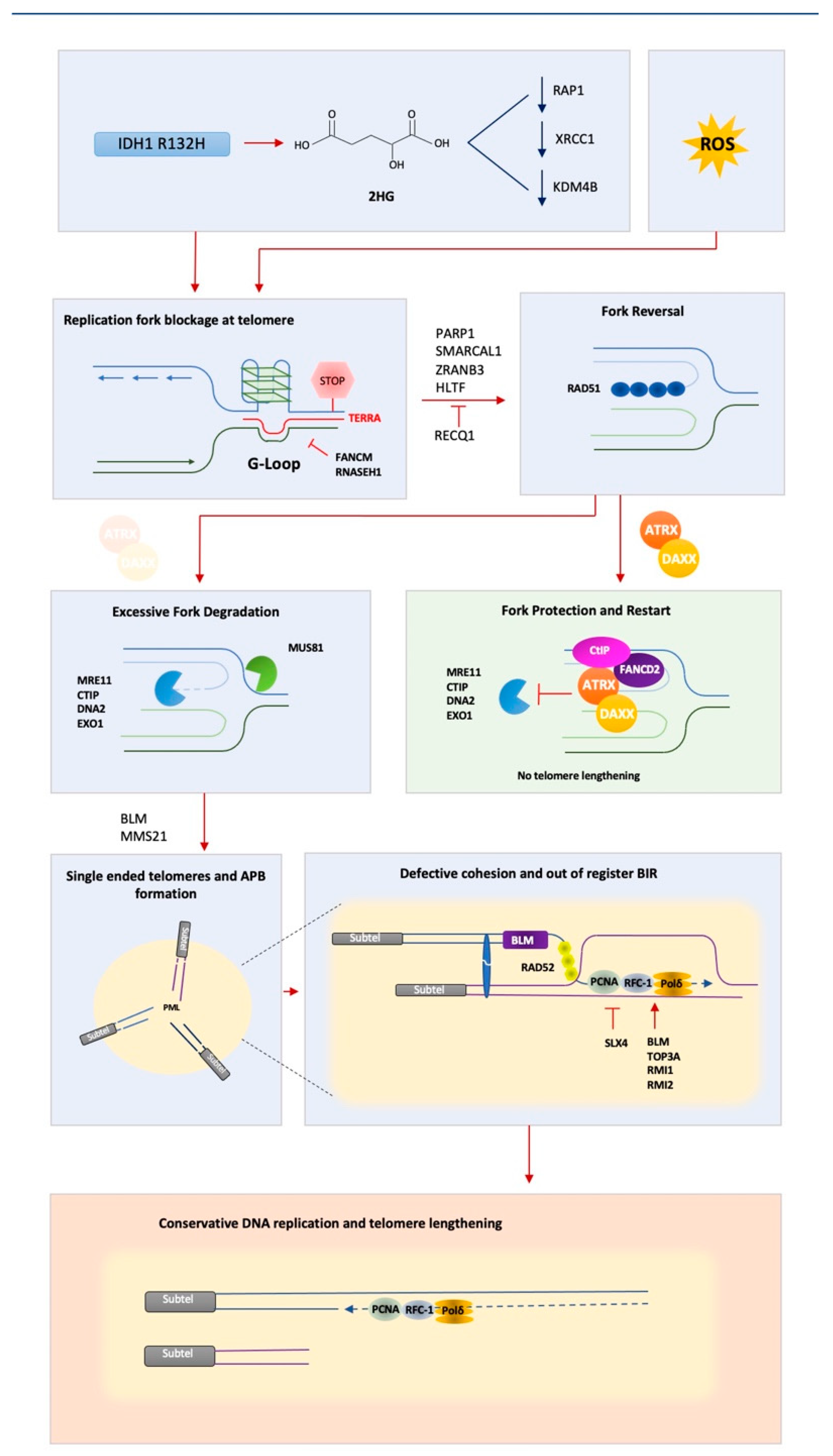
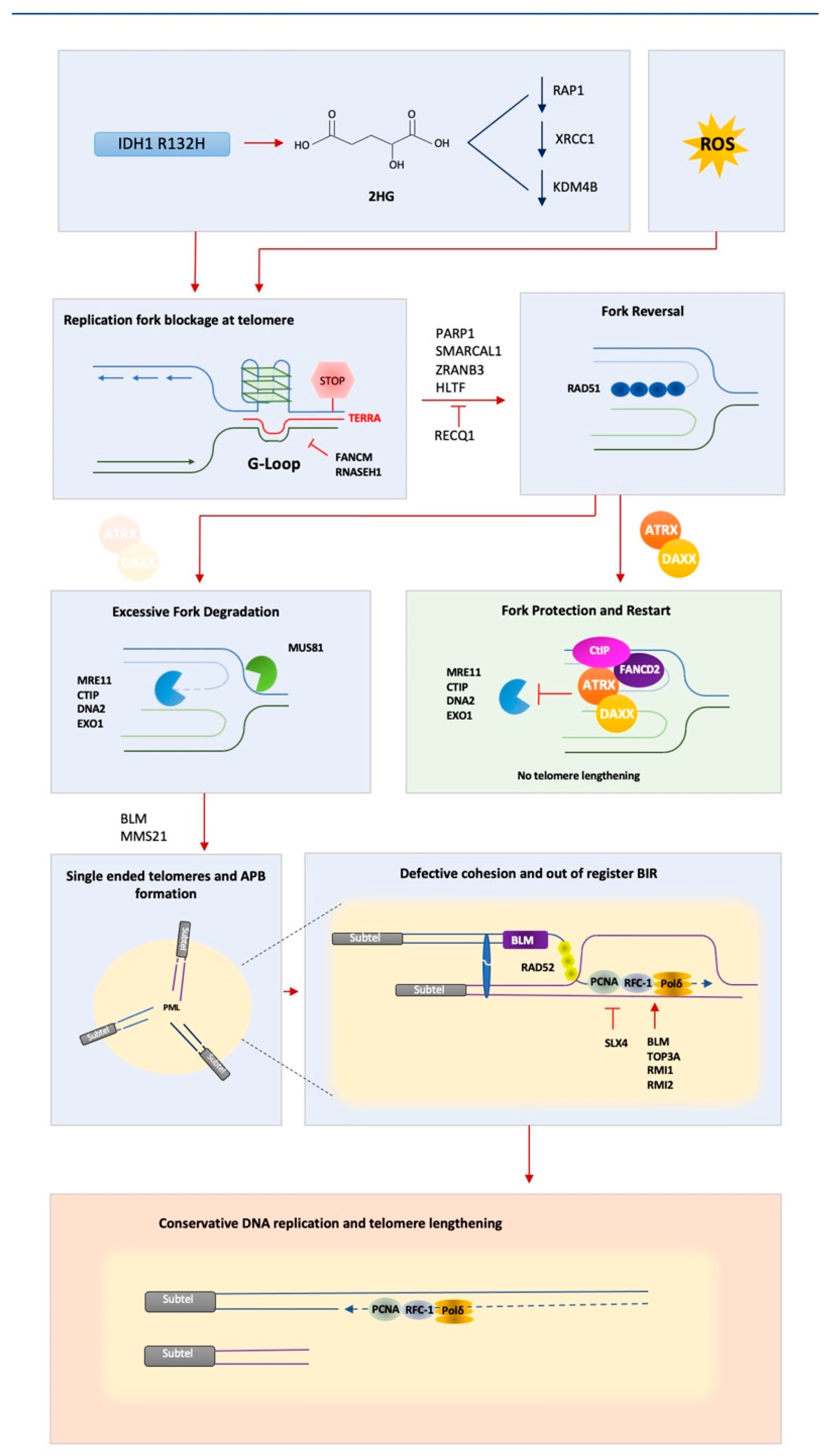
5. Replicative Stress at Telomeres—A Problem on Repeat
6. ATRX—A Key Player in Suppressing ALT
7. Closing Thoughts
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Sotiriou, S.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Ré, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-Induced Replication Repair of Damaged Forks Induces Genomic Duplications in Human Cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Saini, N.; Ramakrishnan, S.; Elango, R.; Ayyar, S.; Zhang, Y.; Deem, A.; Ira, G.; Haber, J.E.; Lobachev, K.S.; Malkova, A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nat. Cell Biol. 2013, 502, 389–392. [Google Scholar] [CrossRef] [Green Version]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nat. Cell Biol. 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, A.; Garcia-Muse, T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- De Lange, T. How Telomeres Solve the End-Protection Problem Downloaded From. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian Telomeres Resemble Fragile Sites and Require TRF1 for Efficient Replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Takai, K.K.; Lovejoy, C.A.; de Lange, T. Break-induced replication promotes fragile telomere formation. Genes Dev. 2020, 34, 1392–1405. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.-P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Olovnikov, A.M. A Theory of Marginotomy The Incomplete Copying of Template Margin in Enzymic Synthesis of Polym&otides and Biological Significance of the Phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [PubMed]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef] [Green Version]
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425. [Google Scholar] [CrossRef] [Green Version]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.-M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.; Westra, W.H.; et al. Prevalence of the Alternative Lengthening of Telomeres Telomere Maintenance Mechanism in Human Cancer Subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative Lengthening of Telomeres in Mammalian Cells. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- McEachern, M.J.; Haber, J.E. Break-Induced Replication and Recombinational Telomere Elongation in Yeast. Annu. Rev. Biochem. 2006, 75, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues est1− senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef]
- Teng, S.-C.; Zakian, V.A. Telomere-Telomere Recombination Is an Efficient Bypass Pathway for Telomere Maintenance in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 8083–8093. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Ijpma, A.; Greider, C.W. Two Survivor Pathways That Allow Growth in the Absence of Telomerase Are Generated by Distinct Telomere Recombination Events. Mol. Cell. Biol. 2001, 21, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- Le, S.; Moore, J.K.; Haber, J.; Greider, C.W. RAD50 and RAD51 Define Two Pathways That Collaborate to Maintain Telomeres in the Absence of Telomerase. Genet. 1999, 152, 143–152. [Google Scholar] [CrossRef]
- Teng, S.-C.; Chang, J.; McCowan, B.; A Zakian, V. Telomerase-Independent Lengthening of Yeast Telomeres Occurs by an Abrupt Rad50p-Dependent, Rif-Inhibited Recombinational Process. Mol. Cell 2000, 6, 947–952. [Google Scholar] [CrossRef]
- Kockler, Z.W.; Comeron, J.M.; Malkova, A. A unified alternative telomere-lengthening pathway in yeast survivor cells. Mol. Cell 2021, 81, 1816–1829.e5. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell. Biol. 2017, 37, e00226-17. [Google Scholar] [CrossRef] [Green Version]
- Roumelioti, F.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D.; Gagos, S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Maric, M.; Hewitt, G.; Mason-Osann, E.; Gali, H.; Dai, A.; Labadorf, A.; Guervilly, J.-H.; Ruis, P.; Segura-Bayona, S.; et al. SLX4IP Antagonizes Promiscuous BLM Activity during ALT Maintenance. Mol. Cell 2019, 76, 27–43.e11. [Google Scholar] [CrossRef] [Green Version]
- Sobinoff, A.; Allen, J.A.; A Neumann, A.; Yang, S.F.; E Walsh, M.; Henson, J.; Reddel, R.R.; A Pickett, H. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Dilley, R.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, N.W.; Dilley, R.; Lampson, M.A.; Greenberg, R.A. Interchromosomal Homology Searches Drive Directional ALT Telomere Movement and Synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Gonzalez, J.; García-Expósito, L.; Hoang, S.M.; Lynskey, M.L.; Roncaioli, J.L.; Ghosh, A.; Wallace, C.T.; de Vitis, M.; Modesti, M.; Bernstein, K.A.; et al. RAD51AP1 Is an Essential Mediator of Alternative Lengthening of Telomeres. Mol. Cell 2019, 76, 11–26.e7. [Google Scholar] [CrossRef]
- Chung, I.; Osterwald, S.; Deeg, K.I.; Rippe, K. PML Body Meets Telomere: The Beginning of an ALTernate Ending? Nucleus 2012, 3, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhao, R.; Tones, J.; Liu, M.; Dilley, R.; Chenoweth, D.M.; Greenberg, R.A.; Lampson, M.A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell 2020, 31, 2048–2056. [Google Scholar] [CrossRef]
- Min, J.; Wright, W.E.; Shay, J.W. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019, 33, 814–827. [Google Scholar] [CrossRef]
- Lang, M.; Jegou, T.; Chung, I.; Richter, K.; Münch, S.; Udvarhelyi, A.; Cremer, C.; Hemmerich, P.; Engelhardt, J.; Hell, S.W.; et al. Three-dimensional organization of promyelocytic leukemia nuclear bodies. J. Cell Sci. 2010, 123, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Lallemand-Breitenbach, V.; de Thé, H. PML Nuclear Bodies. Cold Spring Harb. Perspect. Biology. 2010, 2, 661. [Google Scholar] [CrossRef]
- Boisvert, F.-M.; Hendzel, M.; Bazett-Jones, D.P. Promyelocytic Leukemia (Pml) Nuclear Bodies Are Protein Structures That Do Not Accumulate RNA. J. Cell Biol. 2000, 148, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Shay, J.W. Telomere clustering drives ALT. Aging 2019, 11, 8046–8047. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar]
- Wu, G.; Jiang, X.; Lee, W.-H.; Chen, P.-L. Assembly of functional ALT-associated promyelocytic leukemia bodies requires Nijmegen Breakage Syndrome 1. Cancer Res. 2003, 63. [Google Scholar]
- Zeng, S.; Xiang, T.; Pandita, T.K.; Gonzalez-Suarez, I.; Gonzalo, S.; Harris, C.C.; Yang, Q. Telomere recombination requires the MUS81 endonuclease. Nat. Cell Biol. 2009, 11, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavropoulos, D.J.; Bradshaw, P.S.; Li, X.; Pasic, I.; Truong, K.; Ikura, M.; Ungrin, M.; Meyn, M.S. The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum. Mol. Genet. 2002, 11, 3135–3144. [Google Scholar] [CrossRef] [Green Version]
- Silva, B.; Pentz, R.; Figueira, A.M.; Arora, R.; Lee, Y.W.; Hodson, C.; Wischnewski, H.; Deans, A.J.; Azzalin, C.M. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Cox, K.E.; Maréchal, A.; Flynn, R.L. SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep. 2016, 14, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Root, H.; Larsen, A.; Komosa, M.; Al-Azri, F.; Li, R.; Bazett-Jones, D.P.; Meyn, M.S. FANCD2 limits BLM-dependent telomere instability in the alternative lengthening of telomeres pathway. Hum. Mol. Genet. 2016, 25, 3255–3268. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; O’Rourke, J.; Sobinoff, A.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Potts, P.R.; Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Genois, M.-M.; Ouyang, J.; Lan, L.; Zou, L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell 2021, 81, 1027–1042.e4. [Google Scholar] [CrossRef] [PubMed]
- Mazzucco, G.; Huda, A.; Galli, M.; Piccini, D.; Giannattasio, M.; Pessina, F.; Doksani, Y. Telomere damage induces internal loops that generate telomeric circles. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Bétous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Poole, L.A.; Zhao, R.; Glick, G.G.; Lovejoy, C.A.; Eischen, C.M.; Cortez, D. SMARCAL1 maintains telomere integrity during DNA replication. Proc. Natl. Acad. Sci. USA 2015, 112, 14864–14869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason-Osann, E.; Dai, A.; Floro, J.; Lock, Y.J.; Reiss, M.; Gali, H.; Matschulat, A.; Labadorf, A.; Flynn, R.L. Identification of a novel gene fusion in ALT positive osteosarcoma. Oncotarget 2018, 9, 32868–32880. [Google Scholar] [CrossRef] [Green Version]
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.; Killela, P.J.; Loriaux, D.B.; Lipp, E.; et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Leman, A.R.; Noguchi, E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle 2012, 11, 3945–3955. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Y.; Chang, E.Y.C.; Lim, J.; Kwan, H.H.; Monchaud, D.; Yip, S.; Stirling, P.C.; Wong, J.M.Y. G-quadruplexes mark alternative lengthening of telomeres. NAR Cancer 2021, 3, zcab031. [Google Scholar] [CrossRef]
- Amato, R.; Valenzuela, M.; Berardinelli, F.; Salvati, E.; Maresca, C.; Leone, S.; Antoccia, A.; Sgura, A. G-quadruplex Stabilization Fuels the ALT Pathway in ALT-positive Osteosarcoma Cells. Genes 2020, 11, 304. [Google Scholar] [CrossRef] [Green Version]
- Feretzaki, M.; Pospisilova, M.; Fernandes, R.V.; Lunardi, T.; Krejci, L.; Lingner, J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nat. Cell Biol. 2020, 587, 303–308. [Google Scholar] [CrossRef]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londoño-Vallejo, A.; Decottignies, A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef]
- Tan, J.; Duan, M.; Yadav, T.; Phoon, L.; Wang, X.; Zhang, J.-M.; Zou, L.; Lan, L. An R-loop-initiated CSB–RAD52–POLD3 pathway suppresses ROS-induced telomeric DNA breaks. Nucleic Acids Res. 2020, 48, 1285–1300. [Google Scholar] [CrossRef]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase Therapy Provokes ALT and Mitochondrial Adaptive Mechanisms in Cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; De Lange, T.; De, S.; Petrini, J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.S.; Higgs, D.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX Represses Alternative Lengthening of Telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef] [Green Version]
- Clynes, D.; Higgs, D.; Gibbons, R. The chromatin remodeller ATRX: A repeat offender in human disease. Trends Biochem. Sci. 2013, 38, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Drané, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.; Banaszynski, L.A.; Noh, K.-M.; Lewis, P.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct Factors Control Histone Variant H3.3 Localization at Specific Genomic Regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H.; McGhie, J.D.; Sim, M.; Anderson, M.A.; Ahn, S.; Hannan, R.; George, A.; Morgan, K.A.; Mann, J.R.; Choo, K.A. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010, 20, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.-C.; Sundaresan, A.; O’Hara, R.; Gant, V.U.; Li, M.; Martire, S.; Warshaw, J.N.; Basu, A.; Banaszynski, L.A. ATRX promotes heterochromatin formation to protect cells from G-quadruplex DNA-mediated stress. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Voon, H.; Xella, B.; Scott, C.; Clynes, D.; Babbs, C.; Ayyub, H.; Kerry, J.; A Sharpe, J.; A Sloane-Stanley, J.; et al. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017, 18, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Huh, M.S.; Ivanochko, D.; Hashem, L.; Curtin, M.; Delorme, M.; Goodall, E.; Yan, K.; Picketts, D.J. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 2016, 7, e2220. [Google Scholar] [CrossRef] [Green Version]
- Leung, W.C.; Ghosal, G.; Wang, W.; Shen, X.; Wang, J.; Li, L.; Chen, J. Alpha Thalassemia/Mental Retardation Syndrome X-linked Gene Product ATRX Is Required for Proper Replication Restart and Cellular Resistance to Replication Stress. J. Biol. Chem. 2013, 288, 6342–6350. [Google Scholar] [CrossRef] [Green Version]
- Watson, L.A.; Solomon, L.A.; Li, J.R.; Jiang, Y.; Edwards, M.; Shin-Ya, K.; Beier, F.; Bérubé, N.G. Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J. Clin. Investig. 2013, 123, 2049–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Taylor, S.; Mitson, M.; Bachrati, C.Z.; Higgs, D.; Gibbons, R.J. ATRX Dysfunction Induces Replication Defects in Primary Mouse Cells. PLoS ONE 2014, 9, e92915. [Google Scholar] [CrossRef] [Green Version]
- Pladevall-Morera, D.; Munk, S.; Ingham, A.; Garribba, L.; Albers, E.; Liu, Y.; Olsen, J.; Lopez-Contreras, A.J. Proteomic characterization of chromosomal common fragile site (CFS)-associated proteins uncovers ATRX as a regulator of CFS stability. Nucleic Acids Res. 2019, 47, 8004–8018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghunandan, M.; Yeo, J.E.; Walter, R.; Saito, K.; Harvey, A.J.; Ittershagen, S.; Lee, E.-A.; Yang, J.; E Hoatlin, M.; Bielinsky, A.K.; et al. Functional cross talk between the Fanconi anemia and ATRX/DAXX histone chaperone pathways promotes replication fork recovery. Hum. Mol. Genet. 2019, 29, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Juhász, S.; Elbakry, A.; Mathes, A.; Löbrich, M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell 2018, 71, 11–24.e7. [Google Scholar] [CrossRef] [Green Version]
- Koschmann, C.; Calinescu, A.-A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. C A N C E R ATRX Loss Promotes Tumor Growth and Impairs Nonhomologous End Joining DNA Repair in Glioma. Sci. Transl. Med. 2016, 8. undefined. [Google Scholar] [CrossRef] [Green Version]
- Eid, R.; Demattei, M.-V.; Episkopou, H.; Gouillou, C.A.; Decottignies, A.; Grandin, N.; Charbonneau, M. Genetic Inactivation ofATRXLeads to a Decrease in the Amount of Telomeric Cohesin and Level of Telomere Transcription in Human Glioma Cells. Mol. Cell. Biol. 2015, 35, 2818–2830. [Google Scholar] [CrossRef] [Green Version]
- Brosnan-Cashman, J.A.; Yuan, M.; Graham, M.K.; Rizzo, A.J.; Myers, K.M.; Davis, C.; Zhang, R.; Esopi, D.M.; Raabe, E.H.; Eberhart, C.G.; et al. ATRX loss induces multiple hallmarks of the alternative lengthening of telomeres (ALT) phenotype in human glioma cell lines in a cell line-specific manner. PLoS ONE 2018, 13, e0204159. [Google Scholar] [CrossRef] [Green Version]
- Fontebasso, A.M.; Schwartzentruber, J.; Khuong-Quang, D.-A.; Liu, X.-Y.; Sturm, D.; Korshunov, A.; Jones, D.T.W.; Witt, H.; Kool, M.; Albrecht, S.; et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013, 125, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, J.; Johannessen, T.-C.A.; Ohba, S.; Chow, T.T.; Jones, L.; Pandita, A.; Pieper, R.O. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Knobbe, C.B.; Munger, J.; Lind, E.F.; Brenner, D.; Bruestle, A.; Harris, I.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nat. Cell Biol. 2012, 488, 656–659. [Google Scholar] [CrossRef] [Green Version]
- Titia de Lange, T. Shelterin-Mediated Telomere Protection. Annu.-Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Doksani, Y.; de Lange, T. Telomere-Internal Double-Strand Breaks Are Repaired by Homologous Recombination and PARP1/Lig3-Dependent End-Joining. Cell Rep. 2016, 17, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udugama, M.; Hii, L.; Garvie, A.; Cervini, M.; Vinod, B.; Chan, F.-L.; Das, P.P.; Mann, J.R.; Collas, P.; Voon, H.P.J.; et al. Mutations inhibiting KDM4B drive ALT activation in ATRX-mutated glioblastomas. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; De Lange, T. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol. 2020, 18, e3000594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kent, T.; Clynes, D. Alternative Lengthening of Telomeres: Lessons to Be Learned from Telomeric DNA Double-Strand Break Repair. Genes 2021, 12, 1734. https://doi.org/10.3390/genes12111734
Kent T, Clynes D. Alternative Lengthening of Telomeres: Lessons to Be Learned from Telomeric DNA Double-Strand Break Repair. Genes. 2021; 12(11):1734. https://doi.org/10.3390/genes12111734
Chicago/Turabian StyleKent, Thomas, and David Clynes. 2021. "Alternative Lengthening of Telomeres: Lessons to Be Learned from Telomeric DNA Double-Strand Break Repair" Genes 12, no. 11: 1734. https://doi.org/10.3390/genes12111734
APA StyleKent, T., & Clynes, D. (2021). Alternative Lengthening of Telomeres: Lessons to Be Learned from Telomeric DNA Double-Strand Break Repair. Genes, 12(11), 1734. https://doi.org/10.3390/genes12111734