
Developmental Acquisition of p53 Functions


Abstract
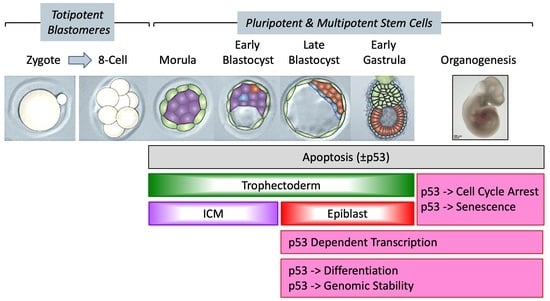
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Current Consensus on p53
2. Current Consensus on Preimplantation Mouse Development
2.1. Transcription of the p53 Gene during Embryonic Development
2.2. p53-Dependent Embryonic Phenotypes
3. What Next?
4. p53 Activity Begins in Pluripotent Stem Cells and Becomes Critical with Gastrulation
4.1. p53 Activity Is First Detected at the Late Blastocyst Stage
4.2. p53 Activity Is Regulated Post-Translationally and Cannot Induce Lethality until Gastrulation
4.3. Take-Home Lesson
5. p53 Maintains Genomic Stability during Embryonic Development
5.1. Tetraploidy -> Polyploidy -> Aneuploidy
5.2. Teratoma Formation
5.3. Take-Home Lesson
6. Maintaining the Pluripotent State
6.1. Pluripotent Stem Cells Maintain p53 at Low Levels
6.2. Pluripotency Is Maintained through p53 Regulation of LncRNA Expression
6.3. Pluripotency Is Maintained through WNT Signaling
6.4. Micro-RNAs Maintain Pluripotency by Inactivating p53
6.5. Pluripotency Is Maintained through p53 Isoforms
6.6. Take-Home Lesson
7. Pluripotent Stem Cell Differentiation
7.1. p53 Facilitates ESC Differentiation
7.2. DNA Damage Induces p53-Dependent Differentiation
7.3. p53 Can Inhibit Pluripotency
7.4. p53 as A Barrier to Reprogramming
7.5. p53-Dependent Formation of Haploid Cells
7.6. Take Home Lesson
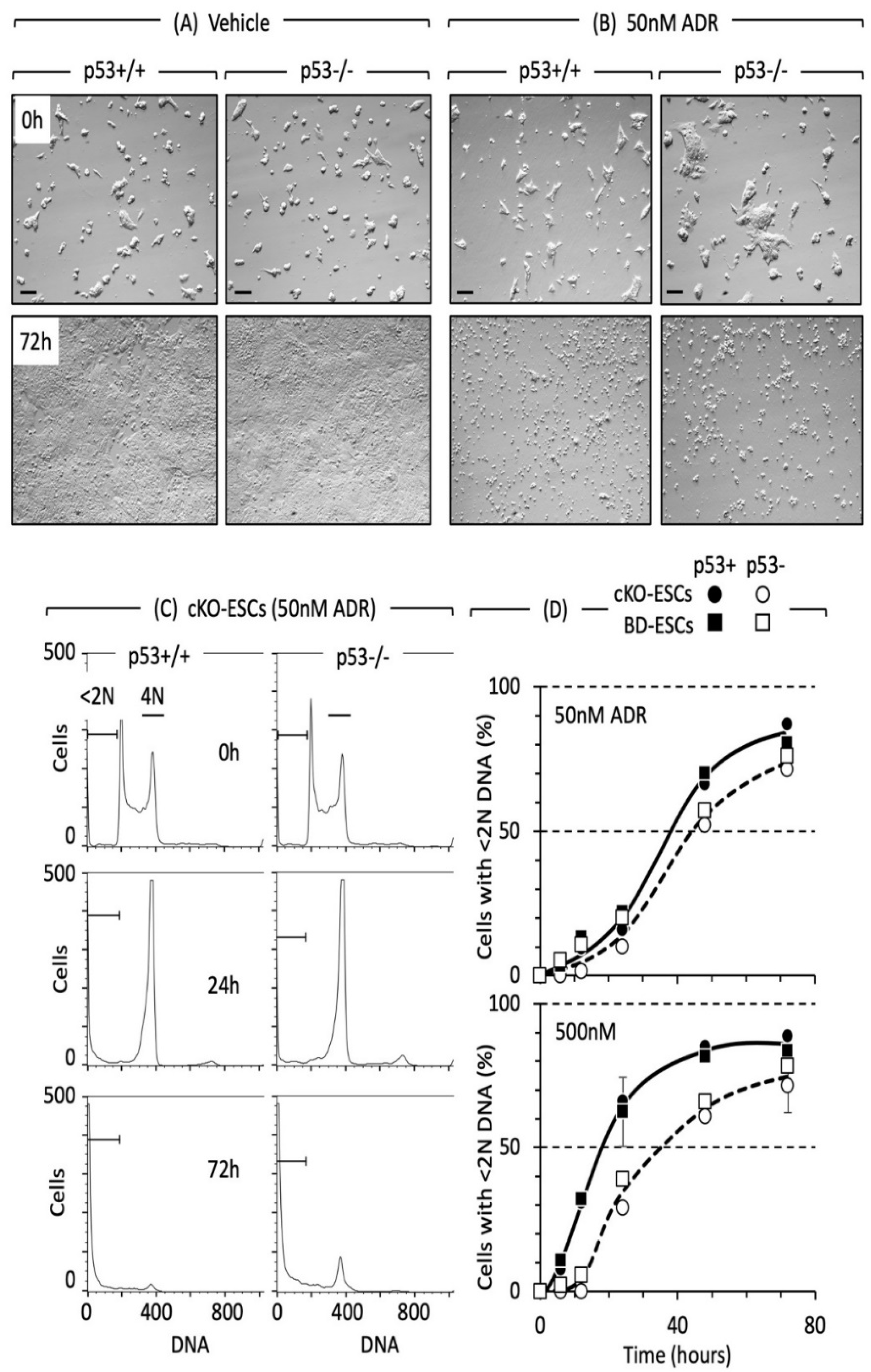
8. DNA Damage Response in ESCs
8.1. Physiological States of Pluripotent Embryonic Stem Cells
8.2. Cell Cycle Arrest in Naïve ESCs Is Not p53-Dependent
8.3. Apoptosis in Naïve ESCs Is Not p53-Dependent
8.4. DNA Damage Response in Ground-State 2iESCs
8.5. Reconciling Disparate Data
8.5.1. Bases for Reconciliation
8.5.2. Reconciliation of Naïve ESC Data
8.5.3. Reconciliation of Ground-State 2iESC Data
8.6. Take-Home Lesson
9. DNA Damage Response in Differentiated Cells
9.1. A p53-Dependent DNA Damage Response Is Acquired during ESC Differentiation
9.2. Viability of Embryonic Fibroblasts Is p53-Independent
9.3. DNA Damage Induces p53-Dependent Senescence in Embryonic Fibroblasts
9.4. Apoptosis Is p53-Dependent in Embryonic Fibroblasts Immortalized with an Oncogene
9.5. Take-Home Lesson
10. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Purdie, C.A.; Harrison, D.J.; Peter, A.; Dobbie, L.; White, S.; Howie, S.E.; Salter, D.M.; Bird, C.C.; Wyllie, A.H.; Hooper, M.L.; et al. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene 1994, 9, 603–609. [Google Scholar]
- Van Nostrand, J.L.; Bowen, M.E.; Vogel, H.; Barna, M.; Attardi, L.D. The p53 family members have distinct roles during mammalian embryonic development. Cell Death Differ. 2017, 24, 575–579. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Berger, S.L. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201. [Google Scholar] [CrossRef]
- Martin-Caballero, J.; Flores, J.M.; Garcia-Palencia, P.; Serrano, M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001, 61, 6234–6238. [Google Scholar] [PubMed]
- Yu, J.; Zhang, L. No PUMA, no death: Implications for p53-dependent apoptosis. Cancer Cell 2003, 4, 248–249. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Barton, M.C. p53: Emerging roles in stem cells, development and beyond. Development 2018, 145, dev158360. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kurabayashi, A.; Shuin, T.; Ohtsuki, Y.; Furihata, M. Overexpression of p53 protein in human tumors. Med. Mol. Morphol. 2012, 45, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Oren, M.; Bartek, J. The sunny side of p53. Cell 2007, 128, 826–828. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Vousden, K.H. p53 regulation of metabolic pathways. Cold Spring Harb. Perspect. Biol. 2010, 2, a001040. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, Y.; Dubois, W.; Wu, X.; Shi, J.; Huang, J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell. 2012, 46, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the cross fire: p53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef]
- Pomerantz, J.H.; Blau, H.M. Tumor suppressors: Enhancers or suppressors of regeneration? Development 2013, 140, 2502–2512. [Google Scholar] [CrossRef]
- Humpton, T.J.; Vousden, K.H. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026146. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Shumate, A.; Pertea, G.; Varabyou, A.; Breitwieser, F.P.; Chang, Y.C.; Madugundu, A.K.; Pandey, A.; Salzberg, S.L. CHESS: A new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018, 19, 208. [Google Scholar] [CrossRef] [PubMed]
- Kunath, T.; Strumpf, D.; Rossant, J. Early trophoblast determination and stem cell maintenance in the mouse—A review. Placenta 2004, 25 (Suppl. A), S32–S38. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.C. How to make a placenta: Mechanisms of trophoblast cell differentiation in mice—A review. Placenta 2005, 26 (Suppl. A), S3–S9. [Google Scholar] [CrossRef]
- Kojima, Y.; Tam, O.H.; Tam, P.P. Timing of developmental events in the early mouse embryo. Semin. Cell Dev. Biol. 2014, 34, 65–75. [Google Scholar] [CrossRef]
- Jurisicova, A.; Latham, K.E.; Casper, R.F.; Casper, R.F.; Varmuza, S.L. Expression and regulation of genes associated with cell death during murine preimplantation embryo development. Mol. Reprod. Dev. 1998, 51, 243–253. [Google Scholar] [CrossRef]
- Wells, D.; Bermudez, M.G.; Steuerwald, N.; Thornhill, A.R.; Walker, D.L.; Malter, H.; Delhanty, J.D.; Cohen, J. Expression of genes regulating chromosome segregation, the cell cycle and apoptosis during human preimplantation development. Hum. Reprod. 2005, 20, 1339–1348. [Google Scholar] [CrossRef]
- Choi, J.; Donehower, L.A. p53 in embryonic development: Maintaining a fine balance. Cell. Mol. Life Sci. 1999, 55, 38–47. [Google Scholar] [CrossRef]
- Tovy, A.; Spiro, A.; McCarthy, R.; Shipony, Z.; Aylon, Y.; Allton, K.; Ainbinder, E.; Furth, N.; Tanay, A.; Barton, M.; et al. p53 is essential for DNA methylation homeostasis in naive embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017, 31, 959–972. [Google Scholar] [CrossRef]
- Shin, M.H.; He, Y.; Huang, J. Embryonic stem cells shed new light on the developmental roles of p53. Cell Biosci. 2013, 3, 42. [Google Scholar] [CrossRef]
- Sah, V.P.; Attardi, L.D.; Mulligan, G.J.; Williams, B.O.; Bronson, R.T.; Jacks, T. A subset of p53-deficient embryos exhibit exencephaly. Nat. Genet. 1995, 10, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.F.; Kaufman, M.H.; Harrison, D.J.; Clarke, A.R. High-frequency developmental abnormalities in p53-deficient mice. Curr. Biol. 1995, 5, 931–936. [Google Scholar] [CrossRef]
- Kaufman, M.H.; Kaufman, D.B.; Brune, R.M.; Stark, M.; Armstrong, J.F.; Clarke, A.R. Analysis of fused maxillary incisor dentition in p53-deficient exencephalic mice. J. Anat. 1997, 191 Pt 1, 57–64. [Google Scholar] [CrossRef]
- Rinon, A.; Molchadsky, A.; Nathan, E.; Yovel, G.; Rotter, V.; Sarig, R.; Tzahor, E. p53 coordinates cranial neural crest cell growth and epithelial-mesenchymal transition/delamination processes. Development 2011, 138, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Atwal, G.S.; Levine, A.J. p53: A new player in reproduction. Cell Cycle 2008, 7, 848–852. [Google Scholar] [CrossRef]
- Goh, A.M.; Lim, C.Y.; Chiam, P.C.; Li, L.; Mann, M.B.; Mann, K.M.; Menendez, S.; Lane, D.P. Using targeted transgenic reporter mice to study promoter-specific p53 transcriptional activity. Proc. Natl. Acad. Sci. USA 2012, 109, 1685–1690. [Google Scholar] [CrossRef]
- Norimura, T.; Nomoto, S.; Katsuki, M.; Gondo, Y.; Kondo, S. p53-dependent apoptosis suppresses radiation-induced teratogenesis. Nat. Med. 1996, 2, 577–580. [Google Scholar] [CrossRef]
- Wilson, Y.; Morris, I.D.; Kimber, S.J.; Brison, D.R. The role of Trp53 in the mouse embryonic response to DNA damage. Mol. Hum. Reprod. 2019, 25, 397–407. [Google Scholar] [CrossRef]
- Solozobova, V.; Blattner, C. Regulation of p53 in embryonic stem cells. Exp. Cell Res. 2010, 316, 2434–2446. [Google Scholar] [CrossRef]
- Solozobova, V.; Rolletschek, A.; Blattner, C. Nuclear accumulation and activation of p53 in embryonic stem cells after DNA damage. BMC Cell Biol. 2009, 10, 46. [Google Scholar] [CrossRef]
- Setoguchi, K.; Teslaa, T.; Koehler, C.M.; Teitell, M.A. p53 Regulates Rapid Apoptosis in Human Pluripotent Stem Cells. J. Mol. Biol. 2016, 428, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Han, M.K.; Song, E.K.; Guo, Y.; Ou, X.; Mantel, C.; Broxmeyer, H.E. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell 2008, 2, 241–251. [Google Scholar] [CrossRef]
- Lee, D.F.; Su, J.; Ang, Y.S.; Carvajal-Vergara, X.; Mulero-Navarro, S.; Pereira, C.F.; Gingold, J.; Wang, H.L.; Zhao, R.; Sevilla, A.; et al. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. Cell Stem Cell 2012, 11, 179–194. [Google Scholar] [CrossRef]
- Maimets, T.; Neganova, I.; Armstrong, L.; Lako, M. Activation of p53 by nutlin leads to rapid differentiation of human embryonic stem cells. Oncogene 2008, 27, 5277–5287. [Google Scholar] [CrossRef]
- Suvorova, I.I.; Grigorash, B.B.; Chuykin, I.A.; Pospelova, T.V.; Pospelov, V.A. G1 checkpoint is compromised in mouse ESCs due to functional uncoupling of p53-p21Waf1 signaling. Cell Cycle 2016, 15, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.M.; de Queiroz, R.M.; Venkatesh, D.; Prives, C. The roles and regulation of MDM2 and MDMX: It is not just about p53. Genes Dev. 2021, 35, 575–601. [Google Scholar] [CrossRef]
- Ivanov, G.S.; Ivanova, T.; Kurash, J.; Ivanov, A.; Chuikov, S.; Gizatullin, F.; Herrera-Medina, E.M.; Rauscher, F., 3rd; Reinberg, D.; Barlev, N.A. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell Biol. 2007, 27, 6756–6769. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Hull, R.; Oosthuysen, B.; Cajee, U.F.; Mokgohloa, L.; Nweke, E.; Antunes, R.J.; Coetzer, T.H.; Ntwasa, M. The Drosophila retinoblastoma binding protein 6 family member has two isoforms and is potentially involved in embryonic patterning. Int. J. Mol. Sci. 2015, 16, 10242–10266. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Deng, B.; Xing, G.; Teng, Y.; Tian, C.; Cheng, X.; Yin, X.; Yang, J.; Gao, X.; Zhu, Y.; et al. PACT is a negative regulator of p53 and essential for cell growth and embryonic development. Proc. Natl. Acad. Sci. USA 2007, 104, 7951–7956. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Q.; Yang, X.; Zhao, M.; Yang, T.; Yao, A.; Tian, X. PACT cessation overcomes ovarian cancer cell chemoresistance to cisplatin by enhancing p53-mediated apoptotic pathway. Biochem. Biophys. Res. Commun. 2019, 511, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Pan, Y.; Christian, J.; Hui, T.; May, W.S., Jr. The RAX/PACT-PKR stress response pathway promotes p53 sumoylation and activation, leading to G(1) arrest. Cell Cycle 2012, 11, 407–417. [Google Scholar] [CrossRef]
- Fujitani, K.; Otomo, A.; Nagayama, Y.; Tachibana, T.; Kato, R.; Kawashima, Y.; Kodera, Y.; Kato, T.; Takada, S.; Tamura, K.; et al. PACT/PRKRA and p53 regulate transcriptional activity of DMRT1. Genet. Mol. Biol. 2020, 43, e20190017. [Google Scholar] [CrossRef]
- Bardot, E.S.; Hadjantonakis, A.K. Mouse gastrulation: Coordination of tissue patterning, specification and diversification of cell fate. Mech. Dev. 2020, 163, 103617. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef]
- Jones, S.N.; Sands, A.T.; Hancock, A.R.; Vogel, H.; Donehower, L.A.; Linke, S.P.; Wahl, G.M.; Bradley, A. The tumorigenic potential and cell growth characteristics of p53-deficient cells are equivalent in the presence or absence of Mdm2. Proc. Natl. Acad. Sci. USA 1996, 93, 14106–14111. [Google Scholar] [CrossRef] [PubMed]
- McMasters, K.M.; Montes de Oca Luna, R.; Pena, J.R.; Lozano, G. mdm2 deletion does not alter growth characteristics of p53-deficient embryo fibroblasts. Oncogene 1996, 13, 1731–1736. [Google Scholar]
- Migliorini, D.; Lazzerini Denchi, E.; Danovi, D.; Jochemsen, A.; Capillo, M.; Gobbi, A.; Helin, K.; Pelicci, P.G.; Marine, J.C. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol. Cell. Biol. 2002, 22, 5527–5538. [Google Scholar] [CrossRef]
- Finch, R.A.; Donoviel, D.B.; Potter, D.; Shi, M.; Fan, A.; Freed, D.D.; Wang, C.Y.; Zambrowicz, B.P.; Ramirez-Solis, R.; Sands, A.T.; et al. mdmx is a negative regulator of p53 activity in vivo. Cancer Res. 2002, 62, 3221–3225. [Google Scholar] [PubMed]
- Parant, J.; Chavez-Reyes, A.; Little, N.A.; Yan, W.; Reinke, V.; Jochemsen, A.G.; Lozano, G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 2001, 29, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Reyes, A.; Parant, J.M.; Amelse, L.L.; de Oca Luna, R.M.; Korsmeyer, S.J.; Lozano, G. Switching mechanisms of cell death in mdm2- and mdm4-null mice by deletion of p53 downstream targets. Cancer Res. 2003, 63, 8664–8669. [Google Scholar] [PubMed]
- Aylon, Y.; Oren, M. p53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef]
- Vassilev, A.; DePamphilis, M.L. Links between DNA Replication, Stem Cells and Cancer. Genes 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H.; Wahl, G.M. p53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res. 1998, 58, 396–401. [Google Scholar]
- Lanni, J.S.; Jacks, T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol. Cell. Biol. 1998, 18, 1055–1064. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Duijf, P.H.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Senovilla, L.; Jemaa, M.; Michaud, M.; Galluzzi, L.; Kepp, O.; Nanty, L.; Criollo, A.; Rello-Varona, S.; Manic, G.; et al. Multipolar mitosis of tetraploid cells: Inhibition by p53 and dependency on Mos. EMBO J. 2010, 29, 1272–1284. [Google Scholar] [CrossRef]
- Li, M.; Fang, X.; Baker, D.J.; Guo, L.; Gao, X.; Wei, Z.; Han, S.; van Deursen, J.M.; Zhang, P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 14188–14193. [Google Scholar] [CrossRef]
- Adler-Wailes, D.C.; Kramer, J.A.; DePamphilis, M.L. Geminin Is Essential for Pluripotent Cell Viability During Teratoma Formation, but Not for Differentiated Cell Viability During Teratoma Expansion. Stem Cells Dev. 2017, 26, 285–302. [Google Scholar] [CrossRef]
- Rivlin, N.; Katz, S.; Doody, M.; Sheffer, M.; Horesh, S.; Molchadsky, A.; Koifman, G.; Shetzer, Y.; Goldfinger, N.; Rotter, V.; et al. Rescue of embryonic stem cells from cellular transformation by proteomic stabilization of mutant p53 and conversion into WT conformation. Proc. Natl. Acad. Sci. USA 2014, 111, 7006–7011. [Google Scholar] [CrossRef] [PubMed]
- Benedict, B.; van Harn, T.; Dekker, M.; Hermsen, S.; Kucukosmanoglu, A.; Pieters, W.; Delzenne-Goette, E.; Dorsman, J.C.; Petermann, E.; Foijer, F.; et al. Loss of p53 suppresses replication-stress-induced DNA breakage in G1/S checkpoint deficient cells. eLife 2018, 7, e37868. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, M.; Ohtsuka, S.; Nishikawa-Torikai, S.; Yamane, M.; Fujii, S.; Murakami, K.; Niwa, H. Maintenance of pluripotency in mouse ES cells without Trp53. Sci. Rep. 2013, 3, 2944. [Google Scholar] [CrossRef]
- Li, Y.; Huang, C.; Zha, L.; Kong, M.; Yang, Q.; Zhu, Y.; Peng, Y.; Ouyang, Q.; Lu, G.; Lin, G.; et al. Generation of NERCe003-A-3, a p53 compound heterozygous mutation human embryonic stem cell line, by CRISPR/Cas9 editing. Stem Cell Res. 2019, 34, 101371. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.; Sands, A.T.; Weiss, R.S.; Hegi, M.E.; Wiseman, R.W.; Pantazis, P.; Giovanella, B.C.; Tainsky, M.A.; Bradley, A.; Donehower, L.A. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene 1993, 8, 2457–2467. [Google Scholar]
- Duensing, A.; Duensing, S. Guilt by association? p53 and the development of aneuploidy in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 694–700. [Google Scholar] [CrossRef]
- Mora, P.T.; Chandrasekaran, K.; McFarland, V.W. An embryo protein induced by SV40 virus transformation of mouse cells. Nature 1980, 288, 722–724. [Google Scholar] [CrossRef]
- Rogel, A.; Popliker, M.; Webb, C.G.; Oren, M. p53 cellular tumor antigen: Analysis of mRNA levels in normal adult tissues, embryos, and tumors. Mol. Cell. Biol. 1985, 5, 2851–2855. [Google Scholar] [CrossRef]
- Schmid, P.; Lorenz, A.; Hameister, H.; Montenarh, M. Expression of p53 during mouse embryogenesis. Development 1991, 113, 857–865. [Google Scholar] [CrossRef]
- Abdelalim, E.M.; Tooyama, I. The p53 inhibitor, pifithrin-α, suppresses self-renewal of embryonic stem cells. Biochem. Biophys. Res. Commun. 2012, 420, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Singh, M.; Selivanova, G.; Peuget, S. Pifithrin-α alters p53 post-translational modifications pattern and differentially inhibits p53 target genes. Sci. Rep. 2020, 10, 1049. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef]
- Cole, M.F.; Johnstone, S.E.; Newman, J.J.; Kagey, M.H.; Young, R.A. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 2008, 22, 746–755. [Google Scholar] [CrossRef]
- Yi, F.; Pereira, L.; Merrill, B.J. Tcf3 functions as a steady-state limiter of transcriptional programs of mouse embryonic stem cell self-renewal. Stem Cells 2008, 26, 1951–1960. [Google Scholar] [CrossRef]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef]
- Ho, J.S.; Ma, W.; Mao, D.Y.; Benchimol, S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol. Cell. Biol. 2005, 25, 7423–7431. [Google Scholar] [CrossRef]
- Jain, A.K.; Barton, M.C. Making sense of ubiquitin ligases that regulate p53. Cancer Biol. Ther. 2010, 10, 665–672. [Google Scholar] [CrossRef]
- Jain, A.K.; Allton, K.; Iacovino, M.; Mahen, E.; Milczarek, R.J.; Zwaka, T.P.; Kyba, M.; Barton, M.C. p53 regulates cell cycle and microRNAs to promote differentiation of human embryonic stem cells. PLoS Biol. 2012, 10, e1001268. [Google Scholar] [CrossRef]
- Zhang, Z.N.; Chung, S.K.; Xu, Z.; Xu, Y. Oct4 maintains the pluripotency of human embryonic stem cells by inactivating p53 through Sirt1-mediated deacetylation. Stem Cells 2014, 32, 157–165. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Rinn, J.L. lncRNAs: Linking RNA to chromatin. Cold Spring Harb. Perspect. Biol. 2014, 6, a018614. [Google Scholar] [CrossRef]
- Jain, A.K.; Xi, Y.; McCarthy, R.; Allton, K.; Akdemir, K.C.; Patel, L.R.; Aronow, B.; Lin, C.; Li, W.; Yang, L.; et al. LncPRESS1 Is a p53-Regulated LncRNA that Safeguards Pluripotency by Disrupting SIRT6-Mediated De-acetylation of Histone H3K56. Mol. Cell 2016, 64, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Song, C.; Young, N.L.; Sperling, A.S.; Xu, F.; Sridharan, R.; Conway, A.E.; Garcia, B.A.; Plath, K.; Clark, A.T.; et al. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol. Cell 2009, 33, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Chang, K.Y.; Li, Z.; Gates, K.; Rana, Z.A.; Dang, J.; Zhang, D.; Han, T.; Yang, C.S.; Cunningham, T.J.; et al. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol. Cell 2014, 53, 1005–1019. [Google Scholar] [CrossRef]
- Lee, K.H.; Li, M.; Michalowski, A.M.; Zhang, X.; Liao, H.; Chen, L.; Xu, Y.; Wu, X.; Huang, J. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Teresky, A.K.; Levine, A.J. p53 regulates maternal reproduction through LIF. Nature 2007, 450, 721–724. [Google Scholar] [CrossRef]
- Dravid, G.; Ye, Z.; Hammond, H.; Chen, G.; Pyle, A.; Donovan, P.; Yu, X.; Cheng, L. Defining the role of Wnt/β-catenin signaling in the survival, proliferation, and self-renewal of human embryonic stem cells. Stem Cells 2005, 23, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Nishinakamura, R.; Iwamatsu, Y.; Shimosato, D.; Niwa, H. Synergistic action of Wnt and LIF in maintaining pluripotency of mouse ES cells. Biochem. Biophys. Res. Commun. 2006, 343, 159–166. [Google Scholar] [CrossRef]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Melton, C.; Li, Y.P.; Shenoy, A.; Zhang, X.X.; Subramanyam, D.; Blelloch, R. miR-294/miR-302 promotes proliferation, suppresses G1-S restriction point, and inhibits ESC differentiation through separable mechanisms. Cell Rep. 2013, 4, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, C.; Skamagki, M.; Khodadadi-Jamayran, A.; Zhang, W.; Kong, D.; Chang, C.W.; Feng, J.; Han, X.; Townes, T.M.; et al. Elevated p53 Activities Restrict Differentiation Potential of MicroRNA-Deficient Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 1604–1617. [Google Scholar] [CrossRef]
- Ungewitter, E.; Scrable, H. Delta40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. 2010, 24, 2408–2419. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Verhaegh, G.; North, S.; Luciani, M.G.; Lassus, P.; Hibner, U.; Oren, M.; Hainaut, P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002, 21, 6722–6728. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhou, W.; Jiang, J.; Chen, X. Identification of a novel p53 functional domain that is necessary for mediating apoptosis. J. Biol. Chem. 1998, 273, 13030–13036. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Stewart, D.; Matlashewski, G. Regulation of human p53 activity and cell localization by alternative splicing. Mol. Cell. Biol. 2004, 24, 7987–7997. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Stewart, M.H.; Menendez, P.; George, D.; Vijayaragavan, K.; Werbowetski-Ogilvie, T.; Ramos-Mejia, V.; Rouleau, A.; Yang, J.; Bosse, M.; et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature 2007, 448, 1015–1021. [Google Scholar] [CrossRef]
- Hanna, J.; Saha, K.; Pando, B.; van Zon, J.; Lengner, C.J.; Creyghton, M.P.; van Oudenaarden, A.; Jaenisch, R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 2009, 462, 595–601. [Google Scholar] [CrossRef]
- Lin, Y.; Cheng, Z.; Yang, Z.; Zheng, J.; Lin, T. DNp73 improves generation efficiency of human induced pluripotent stem cells. BMC Cell Biol. 2012, 13, 9. [Google Scholar] [CrossRef]
- Fatt, M.P.; Cancino, G.I.; Miller, F.D.; Kaplan, D.R. p63 and p73 coordinate p53 function to determine the balance between survival, cell death, and senescence in adult neural precursor cells. Cell Death Differ. 2014, 21, 1546–1559. [Google Scholar] [CrossRef][Green Version]
- Zhao, Y.; Yin, X.; Qin, H.; Zhu, F.; Liu, H.; Yang, W.; Zhang, Q.; Xiang, C.; Hou, P.; Song, Z.; et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 2008, 3, 475–479. [Google Scholar] [CrossRef]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Izpisua Belmonte, J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.K.; Oh, J.J.; DePamphilis, M.L. Cell cycle arrest and apoptosis are not dependent on p53 prior to p53-dependent embryonic stem cell differentiation. Stem Cells 2020, 38, 1091–1106. [Google Scholar] [CrossRef]
- Keller, G.M. In vitro differentiation of embryonic stem cells. Curr. Opin. Cell Biol. 1995, 7, 862–869. [Google Scholar] [CrossRef]
- Boheler, K.R.; Czyz, J.; Tweedie, D.; Yang, H.T.; Anisimov, S.V.; Wobus, A.M. Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circ. Res. 2002, 91, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Yu, T.; Qing, T.; Liu, Y.; Zhao, Y.; Cai, J.; Li, J.; Song, Z.; Qu, X.; Zhou, P.; et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J. Biol. Chem. 2007, 282, 5842–5852. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. A new role for p53 in maintaining genetic stability in embryonic stem cells. Cell Cycle 2005, 4, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Hassig, C.A.; Fleischer, T.C.; Billin, A.N.; Schreiber, S.L.; Ayer, D.E. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 1997, 89, 341–347. [Google Scholar] [CrossRef]
- Xin, M.; Davis, C.A.; Molkentin, J.D.; Lien, C.L.; Duncan, S.A.; Richardson, J.A.; Olson, E.N. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc. Natl. Acad. Sci. USA 2006, 103, 11189–11194. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Lowe, S.W. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, M.H. Chromosome analysis of early postimplantation presumptive haploid parthenogenetic mouse embryos. J. Embryol. Exp. Morphol. 1978, 45, 85–91. [Google Scholar]
- Keller, G. Embryonic stem cell differentiation: Emergence of a new era in biology and medicine. Genes Dev. 2005, 19, 1129–1155. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H. Mouse ES cell culture system as a model of development. Dev. Growth Differ. 2010, 52, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Sagi, I.; Chia, G.; Golan-Lev, T.; Peretz, M.; Weissbein, U.; Sui, L.; Sauer, M.V.; Yanuka, O.; Egli, D.; Benvenisty, N. Derivation and differentiation of haploid human embryonic stem cells. Nature 2016, 532, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Samanta, M.; Kalantry, S. Generating primed pluripotent epiblast stem cells: A methodology chapter. Curr. Top. Dev. Biol. 2020, 138, 139–174. [Google Scholar] [CrossRef] [PubMed]
- Mulas, C.; Kalkan, T.; von Meyenn, F.; Leitch, H.G.; Nichols, J.; Smith, A. Defined conditions for propagation and manipulation of mouse embryonic stem cells. Development 2019, 146, dev173146. [Google Scholar] [CrossRef]
- Smith, A. Formative pluripotency: The executive phase in a developmental continuum. Development 2017, 144, 365–373. [Google Scholar] [CrossRef]
- Ying, Q.L.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519–523. [Google Scholar] [CrossRef]
- Alexandrova, S.; Kalkan, T.; Humphreys, P.; Riddell, A.; Scognamiglio, R.; Trumpp, A.; Nichols, J. Selection and dynamics of embryonic stem cell integration into early mouse embryos. Development 2016, 143, 24–34. [Google Scholar] [CrossRef]
- Kojima, Y.; Kaufman-Francis, K.; Studdert, J.B.; Steiner, K.A.; Power, M.D.; Loebel, D.A.; Jones, V.; Hor, A.; de Alencastro, G.; Logan, G.J.; et al. The transcriptional and functional properties of mouse epiblast stem cells resemble the anterior primitive streak. Cell Stem Cell 2014, 14, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Gayen, S.; Maclary, E.; Buttigieg, E.; Hinten, M.; Kalantry, S. A Primary Role for the Tsix lncRNA in Maintaining Random X-Chromosome Inactivation. Cell Rep. 2015, 11, 1251–1265. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.K.; Gayen, S.; Kumar, S.; Maclary, E.; Buttigieg, E.; Hinten, M.; Kumari, A.; Harris, C.; Sado, T.; Kalantry, S. An Xist-activating antisense RNA required for X-chromosome inactivation. Nat. Commun. 2015, 6, 8564. [Google Scholar] [CrossRef]
- Barnaba, N.; LaRocque, J.R. Targeting cell cycle regulation via the G2-M checkpoint for synthetic lethality in melanoma. Cell Cycle 2021, 20, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Taylor, W.R. Analyzing the G2/M checkpoint. Methods Mol. Biol. 2004, 280, 51–82. [Google Scholar] [CrossRef]
- He, H.; Wang, C.; Dai, Q.; Li, F.; Bergholz, J.; Li, Z.; Li, Q.; Xiao, Z.X. p53 and p73 Regulate Apoptosis but Not Cell-Cycle Progression in Mouse Embryonic Stem Cells upon DNA Damage and Differentiation. Stem Cell Rep. 2016, 7, 1087–1098. [Google Scholar] [CrossRef]
- Prost, S.; Bellamy, C.O.; Clarke, A.R.; Wyllie, A.H.; Harrison, D.J. p53-independent DNA repair and cell cycle arrest in embryonic stem cells. FEBS Lett. 1998, 425, 499–504. [Google Scholar] [CrossRef]
- Hume, S.; Dianov, G.L.; Ramadan, K. A unified model for the G1/S cell cycle transition. Nucleic Acids Res. 2020, 48, 12483–12501. [Google Scholar] [CrossRef]
- Aladjem, M.I.; Spike, B.T.; Rodewald, L.W.; Hope, T.J.; Klemm, M.; Jaenisch, R.; Wahl, G.M. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr. Biol. 1998, 8, 145–155. [Google Scholar] [CrossRef]
- Hong, Y.; Stambrook, P.J. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc. Natl. Acad. Sci. USA 2004, 101, 14443–14448. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gou, H.; Tripathi, B.K.; Huang, J.; Jiang, S.; Dubois, W.; Waybright, T.; Lei, M.; Shi, J.; Zhou, M.; et al. An Apela RNA-Containing Negative Feedback Loop Regulates p53-Mediated Apoptosis in Embryonic Stem Cells. Cell Stem Cell 2015, 16, 669–683. [Google Scholar] [CrossRef]
- Ter Huurne, M.; Peng, T.; Yi, G.; van Mierlo, G.; Marks, H.; Stunnenberg, H.G. Critical Role for p53 in Regulating the Cell Cycle of Ground State Embryonic Stem Cells. Stem Cell Rep. 2020, 14, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ter Huurne, M.; Chappell, J.; Dalton, S.; Stunnenberg, H.G. Distinct Cell-Cycle Control in Two Different States of Mouse Pluripotency. Cell Stem Cell 2017, 21, 449–455. [Google Scholar] [CrossRef]
- Corbet, S.W.; Clarke, A.R.; Gledhill, S.; Wyllie, A.H. p53-dependent and -independent links between DNA-damage, apoptosis and mutation frequency in ES cells. Oncogene 1999, 18, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.; Flores, E.R.; Miranda, B.; Hsieh, H.M.; van Oostrom, C.T.; Sage, J.; Jacks, T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc. Natl. Acad. Sci. USA 2002, 99, 2948–2953. [Google Scholar] [CrossRef]
- Grandela, C.; Pera, M.F.; Grimmond, S.M.; Kolle, G.; Wolvetang, E.J. p53 is required for etoposide-induced apoptosis of human embryonic stem cells. Stem Cell Res. 2007, 1, 116–128. [Google Scholar] [CrossRef]
- Atashpaz, S.; Samadi Shams, S.; Gonzalez, J.M.; Sebestyen, E.; Arghavanifard, N.; Gnocchi, A.; Albers, E.; Minardi, S.; Faga, G.; Soffientini, P.; et al. ATR expands embryonic stem cell fate potential in response to replication stress. eLife 2020, 9, e54756. [Google Scholar] [CrossRef] [PubMed]
- Grow, E.J.; Weaver, B.D.; Smith, C.M.; Guo, J.; Stein, P.; Shadle, S.C.; Hendrickson, P.G.; Johnson, N.E.; Butterfield, R.J.; Menafra, R.; et al. p53 convergently activates Dux/DUX4 in embryonic stem cells and in facioscapulohumeral muscular dystrophy cell models. Nat. Genet. 2021, 53, 1207–1220. [Google Scholar] [CrossRef]
- Mansouri, A.; Fukumitsu, H.; Schindehuette, J.; Krieglstein, K. Differentiation of embryonic stem cells. Curr. Protoc. Neurosci. 2009, 47, 3–6. [Google Scholar] [CrossRef]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.; Zambelli, F.; Mertzanidou, A.; Smolders, I.; Geens, M.; Nguyen, H.T.; Barbe, L.; Sermon, K.; Spits, C. Higher-Density Culture in Human Embryonic Stem Cells Results in DNA Damage and Genome Instability. Stem Cell Rep. 2016, 6, 330–341. [Google Scholar] [CrossRef]
- Deng, Y.; Chan, S.S.; Chang, S. Telomere dysfunction and tumour suppression: The senescence connection. Nat. Rev. Cancer 2008, 8, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Cicalese, A.; Fumagalli, M.; Dobreva, M.; Verrecchia, A.; Pelicci, P.G.; di Fagagna, F. DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle 2008, 7, 3601–3606. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: Terminology for TOR-driven aging. Aging 2012, 4, 159–165. [Google Scholar] [CrossRef]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993, 74, 957–967. [Google Scholar] [CrossRef]
- De Rozieres, S.; Maya, R.; Oren, M.; Lozano, G. The loss of mdm2 induces p53-mediated apoptosis. Oncogene 2000, 19, 1691–1697. [Google Scholar] [CrossRef]
- Lowe, S.W.; Bodis, S.; McClatchey, A.; Remington, L.; Ruley, H.E.; Fisher, D.E.; Housman, D.E.; Jacks, T. p53 status and the efficacy of cancer therapy in vivo. Science 1994, 266, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Jacks, T.; Housman, D.E.; Ruley, H.E. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc. Natl. Acad. Sci. USA 1994, 91, 2026–2030. [Google Scholar] [CrossRef]
- Vater, C.A.; Bartle, L.M.; Dionne, C.A.; Littlewood, T.D.; Goldmacher, V.S. Induction of apoptosis by tamoxifen-activation of a p53-estrogen receptor fusion protein expressed in E1A and T24 H-ras transformed p53-/- mouse embryo fibroblasts. Oncogene 1996, 13, 739–748. [Google Scholar] [PubMed]
- Samuelson, A.V.; Lowe, S.W. Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12094–12099. [Google Scholar] [CrossRef]
- Hermeking, H.; Eick, D. Mediation of c-Myc-induced apoptosis by p53. Science 1994, 265, 2091–2093. [Google Scholar] [CrossRef]
- Hosogane, M.; Bosu, L.; Fukumoto, E.; Yamada, H.; Sato, S.; Nakayama, K. Geminin is an indispensable inhibitor of Cdt1 in mouse embryonic stem cells. Genes Cells 2017, 22, 360–375. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Kaneko, K.J.; Pan, H.; DePamphilis, M.L. Geminin is Essential to Prevent DNA Re-Replication-Dependent Apoptosis in Pluripotent Cells, but not in Differentiated Cells. Stem Cells 2015, 33, 3239–3253. [Google Scholar] [CrossRef]
- Iliou, M.S.; Kotantaki, P.; Karamitros, D.; Spella, M.; Taraviras, S.; Lygerou, Z. Reduced Geminin levels promote cellular senescence. Mech. Ageing Dev. 2013, 134, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Pellegata, N.S.; Antoniono, R.J.; Redpath, J.L.; Stanbridge, E.J. DNA damage and p53-mediated cell cycle arrest: A reevaluation. Proc. Natl. Acad. Sci. USA 1996, 93, 15209–15214. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaiswal, S.K.; Raj, S.; DePamphilis, M.L. Developmental Acquisition of p53 Functions. Genes 2021, 12, 1675. https://doi.org/10.3390/genes12111675
Jaiswal SK, Raj S, DePamphilis ML. Developmental Acquisition of p53 Functions. Genes. 2021; 12(11):1675. https://doi.org/10.3390/genes12111675
Chicago/Turabian StyleJaiswal, Sushil K., Sonam Raj, and Melvin L. DePamphilis. 2021. "Developmental Acquisition of p53 Functions" Genes 12, no. 11: 1675. https://doi.org/10.3390/genes12111675
APA StyleJaiswal, S. K., Raj, S., & DePamphilis, M. L. (2021). Developmental Acquisition of p53 Functions. Genes, 12(11), 1675. https://doi.org/10.3390/genes12111675