
Gene Therapies for Monogenic Autism Spectrum Disorders


Abstract
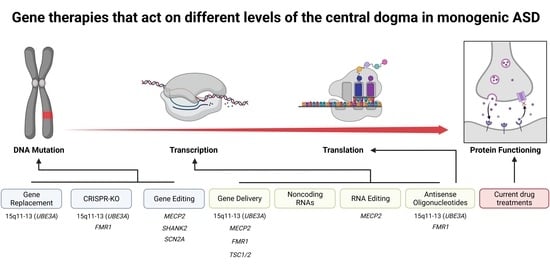
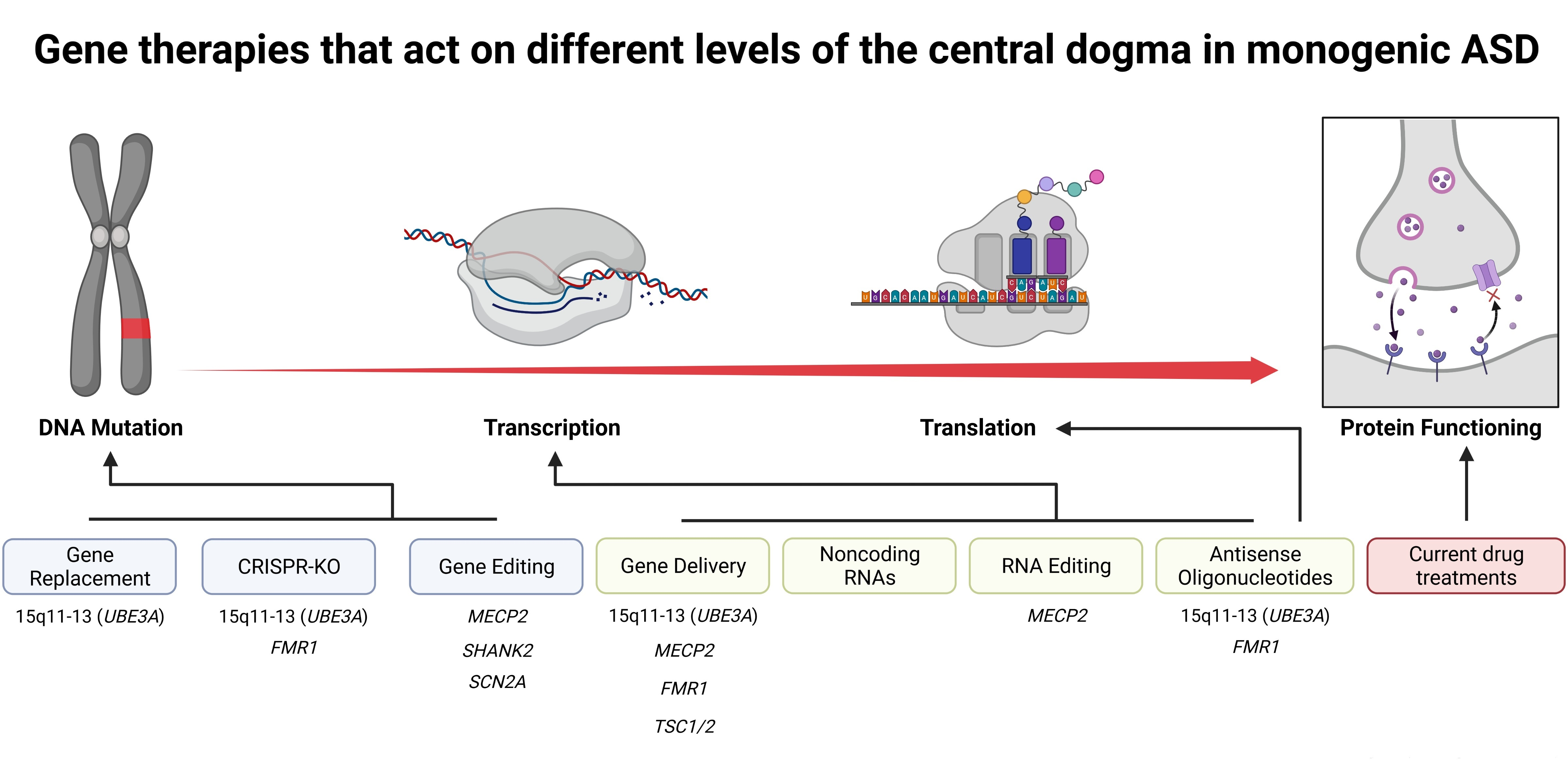
:
1. Introduction
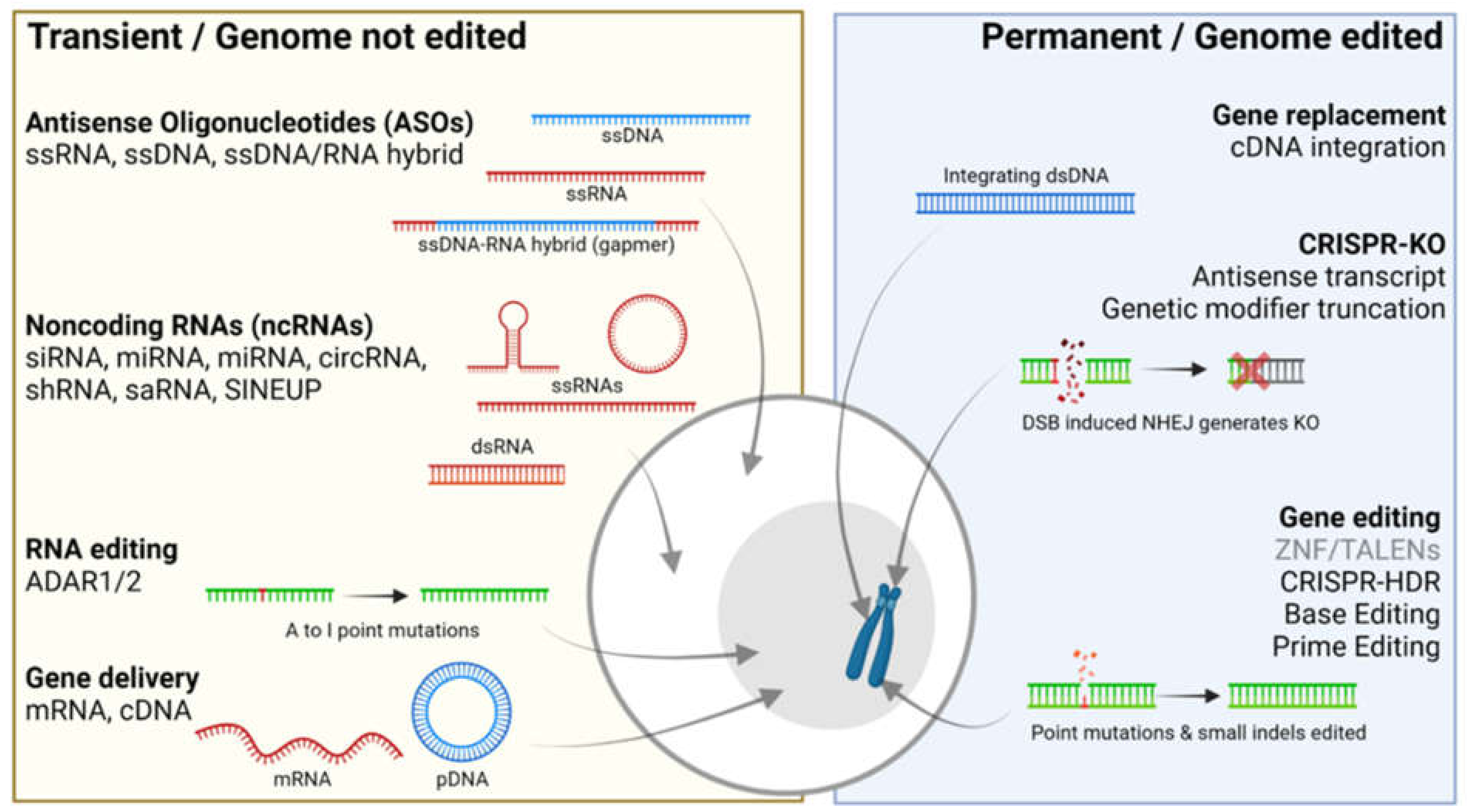
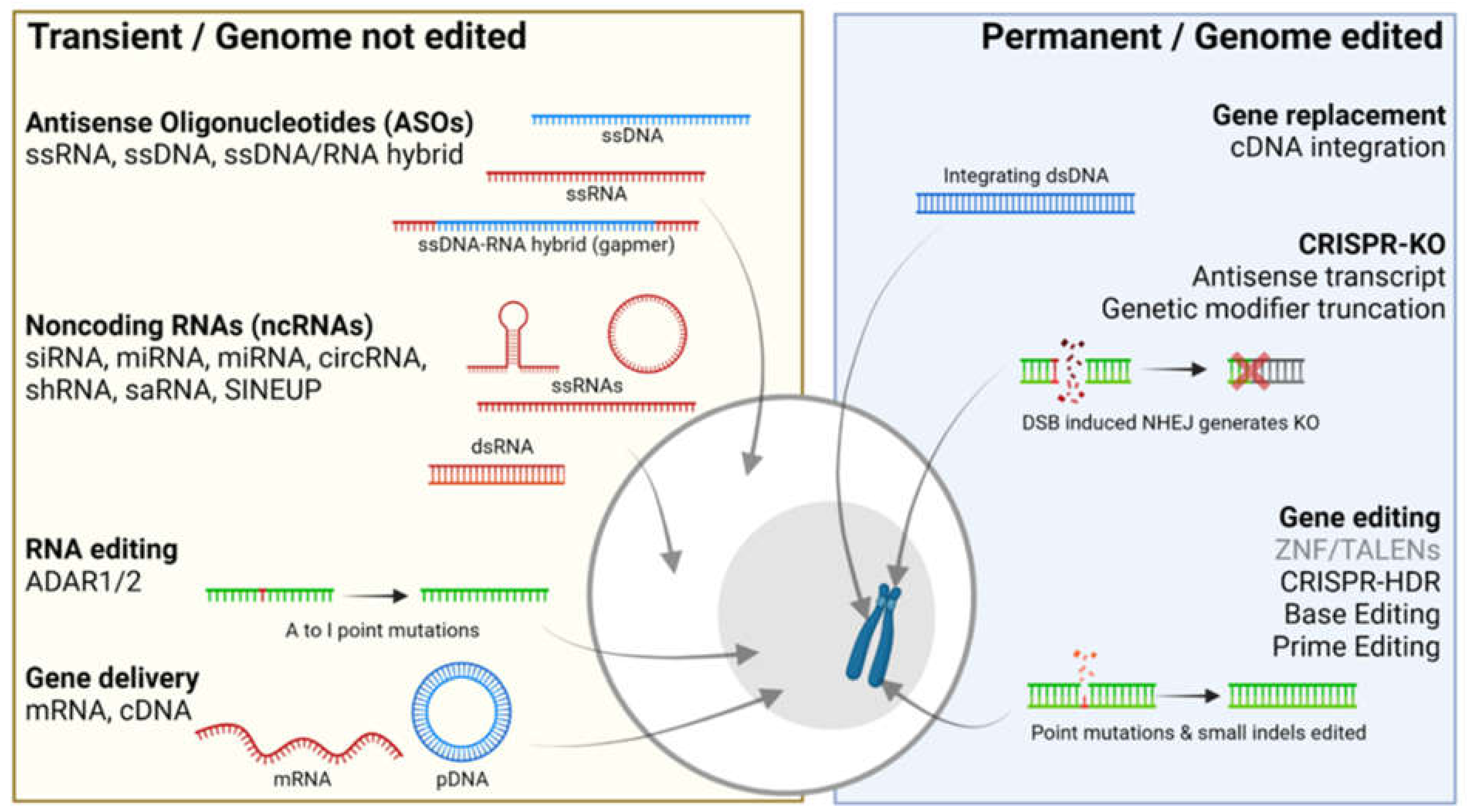
2. Transient Gene Therapies That Do Not Edit the Genome
3. Permanent Gene Therapies That Alter the Genome
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benger, M.; Kinali, M.; Mazarakis, N.D. Autism spectrum disorder: Prospects for treatment using gene therapy. Mol. Autism 2018, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubroth, P.; Colasante, G.; Lignani, G. In vivo Genome Editing Therapeutic Approaches for Neurological Disorders: Where Are We in the Translational Pipeline? Front. Neurosci. 2021, 15, 632522. [Google Scholar] [CrossRef]
- Havdahl, A.; Niarchou, M.; Starnawska, A.; Uddin, M.; van der Merwe, C.; Warrier, V. Genetic contributions to autism spectrum disorder. Psychol. Med. 2021, 51, 2260–2273. [Google Scholar] [CrossRef]
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef]
- Cheroni, C.; Caporale, N.; Testa, G. Autism spectrum disorder at the crossroad between genes and environment: Contributions, convergences, and interactions in ASD developmental pathophysiology. Mol. Autism 2020, 11, 69. [Google Scholar] [CrossRef]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by reduction of a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef]
- Daily, J.L.; Nash, K.; Jinwal, U.; Golde, T.; Rogers, J.; Peters, M.M.; Burdine, R.D.; Dickey, C.; Banko, J.L.; Weeber, E.J. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS ONE 2011, 6, e27221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnamon, J.R.; Kim, S.Y.; Fisk, J.R.; Song, Z.; Nakai, H.; Jeng, S.; McWeeney, S.K.; Mandel, G. In Vivo Repair of a Protein Underlying a Neurological Disorder by Programmable RNA Editing. Cell Rep. 2020, 32, 107878. [Google Scholar] [CrossRef] [PubMed]
- Wolter, J.M.; Mao, H.; Fragola, G.; Simon, J.M.; Krantz, J.L.; Bazick, H.O.; Oztemiz, B.; Stein, J.L.; Zylka, M.J. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature 2020, 587, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Luoni, M.; Giannelli, S.; Indrigo, M.T.; Niro, A.; Massimino, L.; Iannielli, A.; Passeri, L.; Russo, F.; Morabito, G.; Calamita, P.; et al. Whole brain delivery of an instability-prone Mecp2 transgene improves behavioral and molecular pathological defects in mouse models of Rett syndrome. Elife 2020, 9, 1–30. [Google Scholar] [CrossRef]
- Sinnett, S.E.; Hector, R.D.; Gadalla, K.K.E.; Heindel, C.; Chen, D.; Zaric, V.; Bailey, M.E.S.; Cobb, S.R.; Gray, S.J. Improved MECP2 Gene Therapy Extends the Survival of MeCP2-Null Mice without Apparent Toxicity after Intracisternal Delivery. Mol. Ther.-Methods Clin. Dev. 2017, 5, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, A.; Copping, N.A.; Beegle, J.; Cameron, D.L.; Deng, P.; O’Geen, H.; Segal, D.J.; Fink, K.D.; Silverman, J.L.; Anderson, J.S. Functional rescue in an Angelman syndrome model following treatment with lentivector transduced hematopoietic stem cells. Hum. Mol. Genet. 2021, 30, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Huong Le, T.T.; Tran, N.T.; Lan Dao, T.M.; Nguyen, D.D.; Do, H.D.; Ha, T.L.; Kühn, R.; Nguyen, T.L.; Rajewsky, K.; Chu, V.T. Efficient and precise CRISPR/Cas9-mediated MECP2 modifications in human-induced pluripotent stem cells. Front. Genet. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Croci, S.; Carriero, M.L.; Capitani, K.; Daga, S.; Donati, F.; Frullanti, E.; Lamacchia, V.; Tita, R.; Giliberti, A.; Valentino, F.; et al. High rate of HDR in gene editing of p.(Thr158Met) MECP2 mutational hotspot. Eur. J. Hum. Genet. 2020, 28, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Derbis, M.; Kul, E.; Niewiadomska, D.; Sekrecki, M.; Piasecka, A.; Taylor, K.; Hukema, R.K.; Stork, O.; Sobczak, K. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, S.; Arsenault, J.; Xuan, I.C.Y.; Pacey, L.K.; Hampson, D.R. Reduced phenotypic severity following adeno-associated virus-mediated Fmr1 gene delivery in fragile X mice. Neuropsychopharmacology 2014, 39, 3100–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, N.; Gong, H.; Suhl, J.A.; Chopra, P.; Wang, T.; Warren, S.T. Reactivation of FMR1 by CRISPR/Cas9-mediated deletion of the expanded CGG-repeat of the fragile X chromosome. PLoS ONE 2016, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, S.; Cheah, P.S.; Zhang, X.; Zinter, M.; Gianatasio, M.; Hudry, E.; Bronson, R.T.; Kwiatkowski, D.J.; Stemmer-Rachamimov, A.; Maguire, C.A.; et al. Long-Term Therapeutic Efficacy of Intravenous AAV-Mediated Hamartin Replacement in Mouse Model of Tuberous Sclerosis Type 1. Mol. Ther. Methods Clin. Dev. 2019, 15, 18–26. [Google Scholar] [CrossRef]
- Cheah, P.S.; Prabhakar, S.; Yellen, D.; Beauchamp, R.L.; Zhang, X.; Kasamatsu, S.; Bronson, R.T.; Thiele, E.A.; Kwiatkowski, D.J.; Stemmer-Rachamimov, A.; et al. Gene therapy for tuberous sclerosis complex type 2 in a mouse model by delivery of AAV9 encoding a condensed form of tuberin. Sci. Adv. 2021, 7, 1–14. [Google Scholar] [CrossRef]
- Zaslavsky, K.; Zhang, W.B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with Autism Spectrum Disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef]
- Zeliadt, N. CRISPR Therapy May Reverse Autism Mutation’s Effects Well Past Infancy. Spectrum News, 20 October 2019. Available online: https://www.spectrumnews.org/news/crispr-therapy-may-reverse-autism-mutations-effects-well-past-infancy/ (accessed on 15 September 2021).
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J.; et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Chen, X.; Mangala, S.L.; Rodriguez-Aguayo, C.; Kong, X.; Lopez-Berestein, G.; Sood, A.K. RNA interference-based therapy and its delivery systems. Cancer Metastasis Rev. 2018, 37, 107–124. [Google Scholar] [CrossRef]
- Dong, Z.F.; Tang, L.J.; Deng, G.F.; Zeng, T.; Liu, S.J.; Wan, R.P.; Liu, T.; Zhao, Q.H.; Yi, Y.H.; Liao, W.P.; et al. Transcription of the human sodium channel SCN1A gene is repressed by a scaffolding protein RACK1. Mol. Neurobiol. 2014, 50, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Liu, S.J.; Gao, M.M.; Zeng, T.; Lin, G.W.; Tan, N.N.; Tang, H.L.; Lu, P.; Su, T.; Sun, W.W.; et al. MDH2 is an RNA binding protein involved in downregulation of sodium channel Scn1a expression under seizure condition. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.; Raulf, N.; Habib, N. Developing small activating RNA as a therapeutic: Current challenges and promises. Ther. Deliv. 2019, 10, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Kohlmaier, A.; Teupser, D. Circular RNAs as Therapeutic Agents and Targets. Front. Physiol. 2018, 9, 1262. [Google Scholar] [CrossRef] [PubMed]
- Toki, N.; Takahashi, H.; Zucchelli, S.; Gustincich, S.; Carninci, P. Synthetic in vitro transcribed lncRNAs (SINEUPs) with chemical modifications enhance target mRNA translation. FEBS Lett. 2020, 24, 4357–4369. [Google Scholar] [CrossRef]
- Markati, T.; Duis, J.; Servais, L. Therapies in preclinical and clinical development for Angelman syndrome. Expert Opin. Investig. Drugs 2021, 30, 709–720. [Google Scholar] [CrossRef]
- Liang, X.H.; Shen, W.; Sun, H.; Migawa, M.T.; Vickers, T.A.; Crooke, S.T. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat. Biotechnol. 2016, 34, 875–880. [Google Scholar] [CrossRef]
- Stephen, S.L.; Sivanandam, V.G.; Kochanek, S. Homologous and heterologous recombination between adenovirus vector DNA and chromosomal DNA. J. Gene Med. 2008, 10, 1176–1189. [Google Scholar] [CrossRef]
- Hampson, D.R.; Hooper, A.; Niibori, Y. The Application of Adeno-Associated Viral Vector Gene Therapy to the Treatment of Fragile X Syndrome. Brain Sci. 2019, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Merkle, T.; Merz, S.; Reautschnig, P.; Blaha, A.; Li, Q.; Vogel, P.; Wettengel, J.; Li, J.B.; Stafforst, T. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019, 37, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Kafri, T.; Blömer, U.; Peterson, D.A.; Gage, F.H.; Verma, I.M. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat. Genet. 1997, 17, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Schenkwein, D.; Afzal, S.; Nousiainen, A.; Schmidt, M.; Ylä-Herttuala, S. Efficient Nuclease-Directed Integration of Lentivirus Vectors into the Human Ribosomal DNA Locus. Mol. Ther. 2020, 28, 1858–1875. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 1–21. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [Green Version]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Scholefield, J.; Harrison, P.T. Prime editing–an update on the field. Gene Ther. 2021, 28, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Lyu, Q.; Ghanam, A.R.; Lazzarotto, C.R.; Newby, G.A.; Zhang, W.; Choi, M.; Slivano, O.J.; Holden, K.; Walker, J.A.; et al. Prime editing in mice reveals the essentiality of a single base in driving tissue-specific gene expression. Genome Biol. 2021, 22, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chemparathy, A.; Zeng, L.; Kempton, H.R.; Shang, S.; Nakamura, M.; Qi, L.S. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol. Cell 2021, 81, 4333–4345.e4. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Bhattacharjee, O.; Mandal, D.; Sen, M.K.; Dey, D.; Dasgupta, A.; Kazi, T.A.; Gupta, R.; Sinharoy, S.; Acharya, K.; et al. CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life Sci. 2019, 232, 116636. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Ledford, H. CRISPR treatment inserted directly into the body for first time. Nature 2020, 579, 185. [Google Scholar] [CrossRef] [Green Version]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Syndrome | Gene | Patient Mutations | Mouse Model | Transient | Permanent |
|---|---|---|---|---|---|
| Angelman | 15q11-13 (UBE3A) | SV; Loss of allele | Ube3a various | ASO UBE3A-ATS [6] GD rAAV9-Ube3a [7] | GR LV-Ube3a [8] KO Ube3a-ATS [9] |
| Rett | MECP2 | Missense, truncating | Mecp2 various | GD PHP.eB-iMecp2 [10] GD rAAV9-MECP2 [11] RE 317G > A (R106Q) [12] | GE R270X [13] GE T158M [14] |
| Fragile X | FMR1 | Repeat expansion | Fmr1 various | ASO CGG repeat [15] GD rAAV9-FMR1 [16] | KO CGG repeat [17] |
| Tuberous sclerosis | TSC1, TSC2 | Missense, truncating | Tsc1f/f, Tsc2f/− | GD rAAV8/9-TSC1 [18] GD rAAV9-TSC2 [19] | |
| Phelan- McDermid | 22q13 (SHANK3) | SV; Loss of allele | Shank3 various | ||
| Not specified | NLGN4X | Missense, truncating | Nlgn4−/− | ||
| NRXN1A | SV; Large deletions | Nrxn1a−/− | |||
| SHANK2 | Missense, SV | Shank2 various | GE R841C/* [20] | ||
| SCN2A | Missense, truncating, splice-site | Scn2a+/− | GE not specified [21] | ||
| CHD8 | Missense, truncating | Chd8+/∆SL, Chd8+/∆L | |||
| SYNGAP1 | Truncating, splice-site | Syngap1+/− | ASOSYNGAP1 splice-site [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weuring, W.; Geerligs, J.; Koeleman, B.P.C. Gene Therapies for Monogenic Autism Spectrum Disorders. Genes 2021, 12, 1667. https://doi.org/10.3390/genes12111667
Weuring W, Geerligs J, Koeleman BPC. Gene Therapies for Monogenic Autism Spectrum Disorders. Genes. 2021; 12(11):1667. https://doi.org/10.3390/genes12111667
Chicago/Turabian StyleWeuring, Wout, Jeroen Geerligs, and Bobby P. C. Koeleman. 2021. "Gene Therapies for Monogenic Autism Spectrum Disorders" Genes 12, no. 11: 1667. https://doi.org/10.3390/genes12111667
APA StyleWeuring, W., Geerligs, J., & Koeleman, B. P. C. (2021). Gene Therapies for Monogenic Autism Spectrum Disorders. Genes, 12(11), 1667. https://doi.org/10.3390/genes12111667