
Genotyping Analysis by RAD-Seq Reads Is Useful to Assess the Genetic Identity and Relationships of Breeding Lines in Lavender Species Aimed at Managing Plant Variety Protection

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Restriction-Site Associated DNA Sequencing (RAD-Seq) and Data Analysis
2.3. Identification of CDS-Mapping Reads and Reads Related to Terpene and Anthocyanin Biosynthesis Pathways
2.4. DNA Barcoding through Sanger Sequencing for Species Determination
3. Results
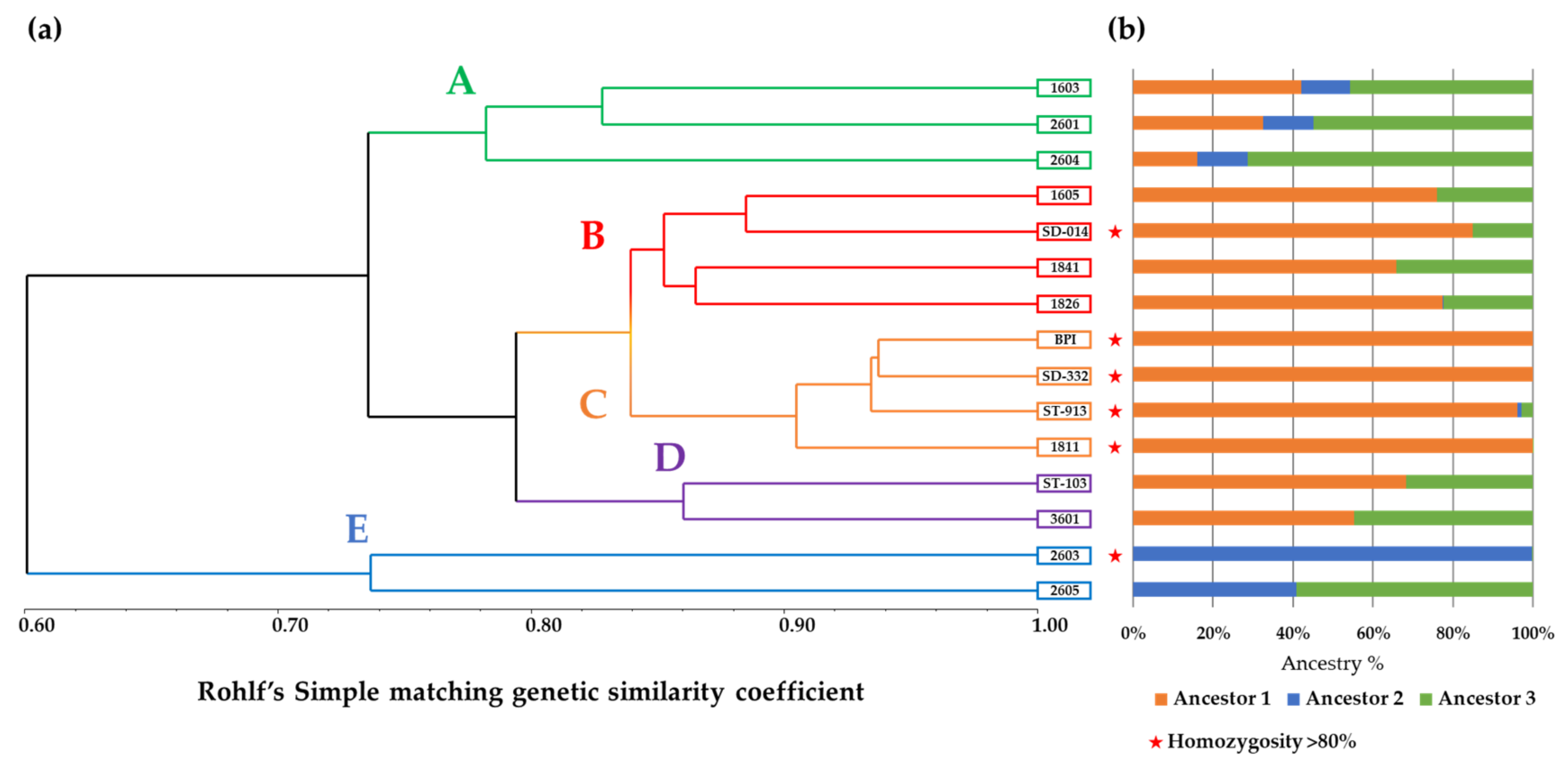
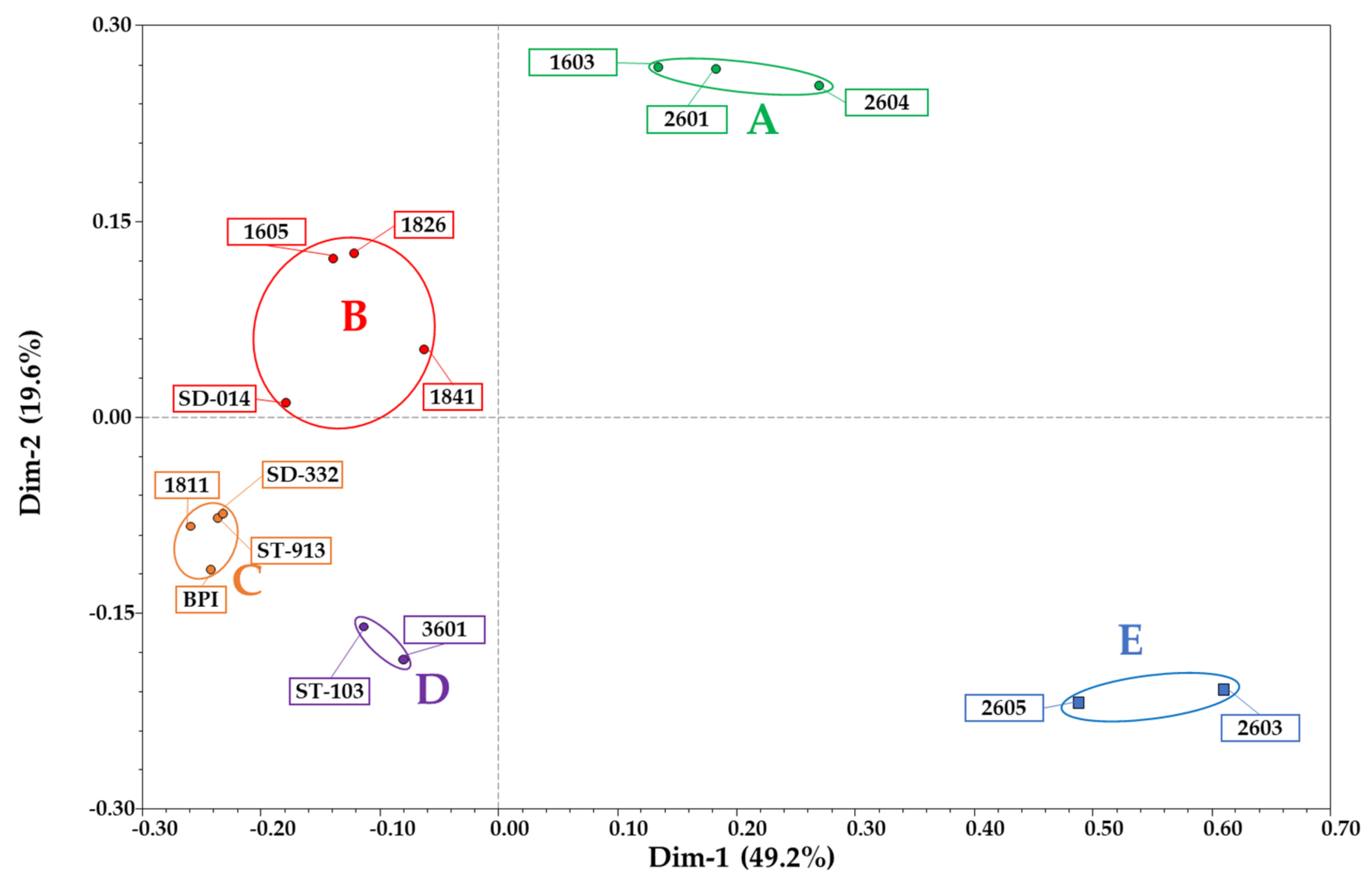
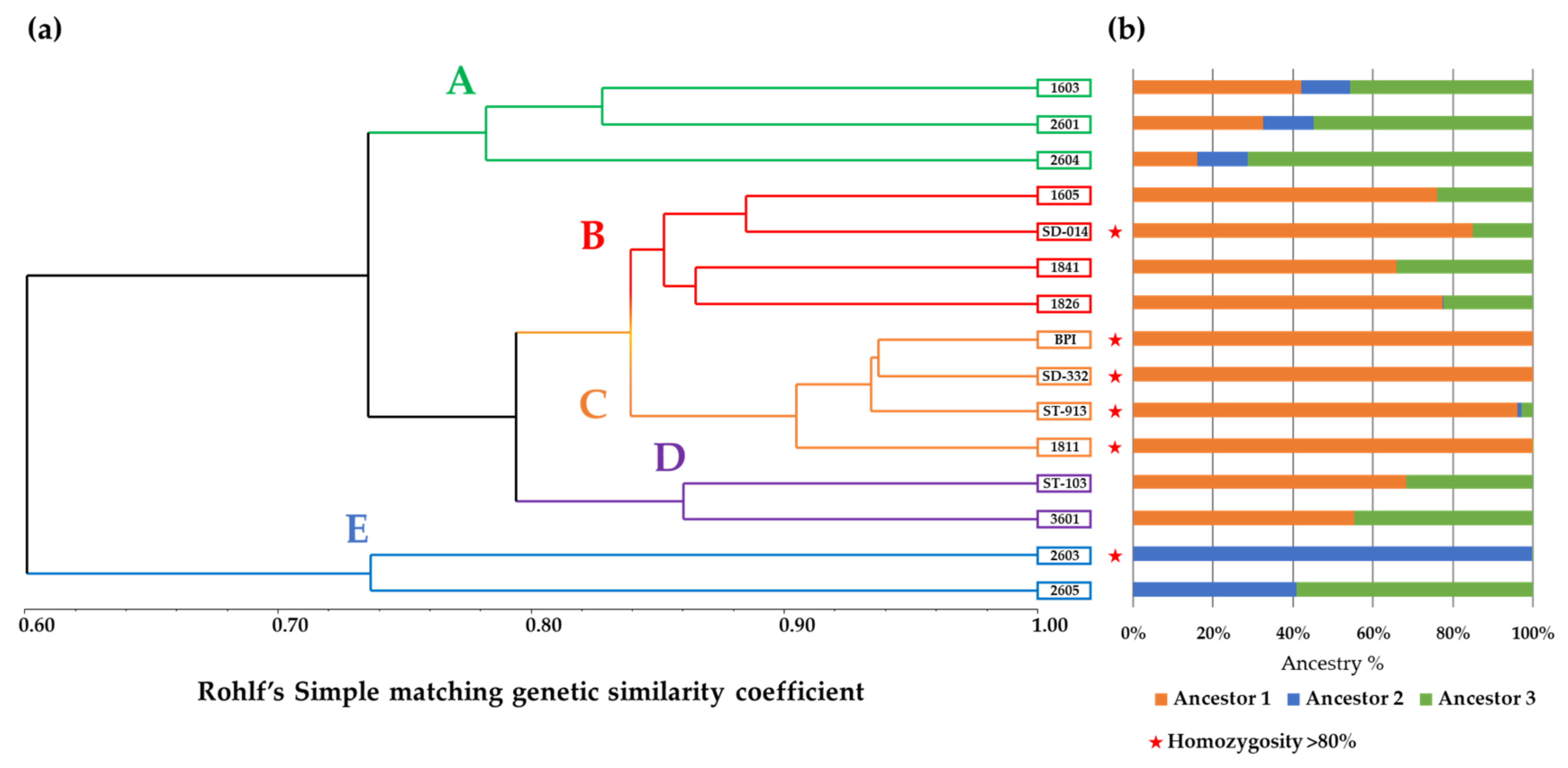
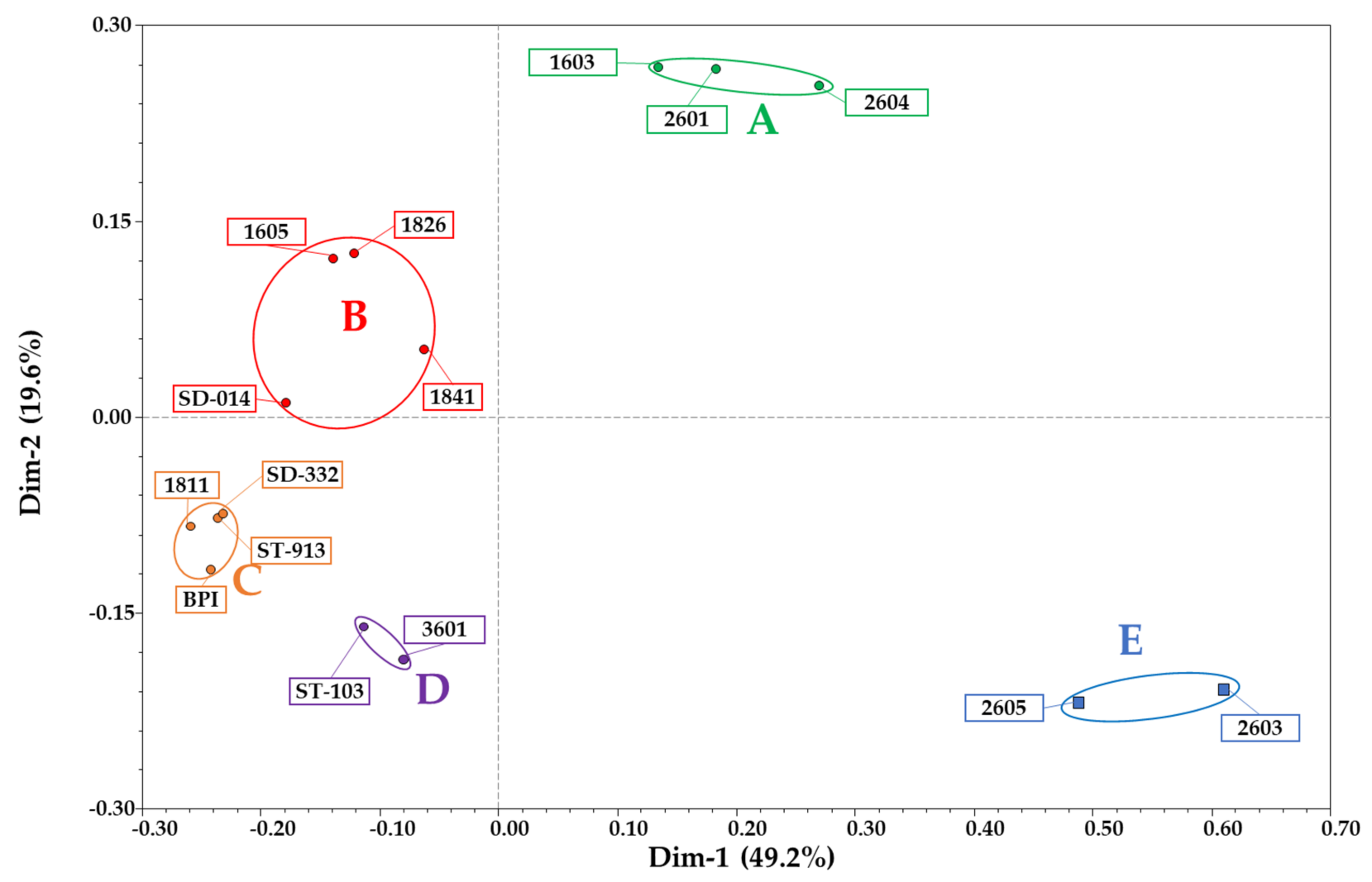
3.1. RAD-Seq and Genetic Similarity Analyses
3.2. CDS-Matching Reads Identification
3.3. BLASTn Analysis for Terpene and Flavonoid Pathway-Related Gene Investigation
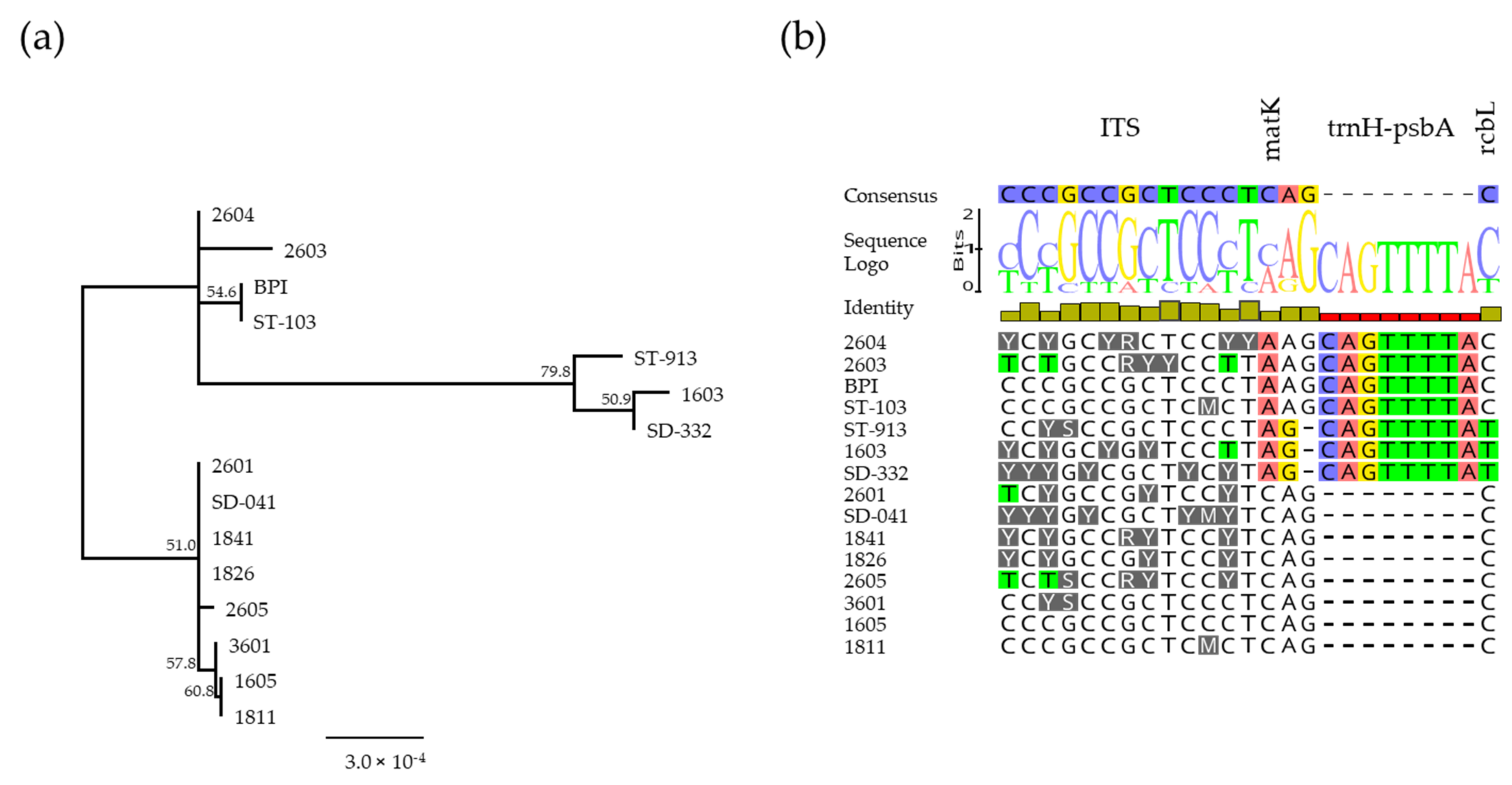
3.4. Sanger Sequencing and DNA Barcoding Analysis
4. Discussion
RAD-Seq-Based Genetic Similarity and Ancestral Composition Reconstruction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rice, A.; Glick, L.; Abadi, S.; Einhorn, M.; Kopelman, N.M.; Salman-Minkov, A.; Mayzel, J.; Chay, O.; Mayrose, I. The Chromosome Counts Database (CCDB)—A community resource of plant chromosome numbers. New Phytol. 2015, 206, 19–26. [Google Scholar] [CrossRef]
- Munoz, A.; Devesa, J. Contribution to the knowledge of the floral biology of the genus Lavandula L., 2: Lavandula stoechas L. subsp. stoechas. An. Jard. Bot. Madr. 1987, 44, 63–78. [Google Scholar]
- Shawl, A.S.; Kumar, S. Potential of lavender oil industry in Kashmir. J. Med. Arom. Plant Sci. 2000, 22, 319–321. [Google Scholar]
- Algieri, F.; Rodriguez-Nogales, A.; Vezza, T.; Garrido-Mesa, J.; Garrido-Mesa, N.; Utrilla, M.P.; Gonzalez-Tejero, M.R.; Casares-Porcel, M.; Molero-Mesa, J.; Del Mar Contreras, M.; et al. Anti-inflammatory activity of hydroalcoholic extracts of Lavandula dentata L. and Lavandula stoechas L. J. Ethnopharmacol. 2016, 190, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Zuzarte, M.d.R. Portuguese Lavenders: Evaluation of Their Potential Use for Health and Agricultural Purposes; Universidade de Coimbra: Coimbra, Portugal, 2013. [Google Scholar]
- Zuzarte, M.; Gonçalves, M.J.; Cavaleiro, C.; Cruz, M.T.; Benzarti, A.; Marongiu, B.; Maxia, A.; Piras, A.; Salgueiro, L. Antifungal and anti-inflammatory potential of Lavandula stoechas and Thymus herba-barona essential oils. Ind. Crop. Prod. 2013, 44, 97–103. [Google Scholar] [CrossRef]
- CPVO. Protocol for Tests on Distinctness, Uniformity and Stability; CPVO, Ed.; Lavandula L., Lavender; European Union: Angers, France, 2018. [Google Scholar]
- Pan, L.; Wang, N.; Wu, Z.; Guo, R.; Yu, X.; Zheng, Y.; Xia, Q.; Gui, S.; Chen, C. A High Density Genetic Map Derived from RAD Sequencing and Its Application in QTL Analysis of Yield-Related Traits in Vigna unguiculata. Front. Plant Sci. 2017, 8, 1544. [Google Scholar] [CrossRef] [Green Version]
- Patella, A.; Palumbo, F.; Ravi, S.; Stevanato, P.; Barcaccia, G. Genotyping by RAD Sequencing Analysis Assessed the Genetic Distinctiveness of Experimental Lines and Narrowed Down the Genomic Region Responsible for Leaf Shape in Endive (Cichorium endivia L.). Genes 2020, 11, 462. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, F.; Galvao, A.C.; Nicoletto, C.; Sambo, P.; Barcaccia, G. Diversity Analysis of Sweet Potato Genetic Resources Using Morphological and Qualitative Traits and Molecular Markers. Genes 2019, 10, 840. [Google Scholar] [CrossRef] [Green Version]
- Barcaccia, G.; Palumbo, F.; Scariolo, F.; Vannozzi, A.; Borin, M.; Bona, S. Potentials and Challenges of Genomics for Breeding Cannabis Cultivars. Front. Plant Sci. 2020, 11, 573299. [Google Scholar] [CrossRef]
- Hnia, C.; Mohamed, B. Genetic diversity of Lavandula multifida L. (Lamiaceae) in Tunisia: Implication for conservation. Afr. J. Ecol. 2011, 49, 10–20. [Google Scholar] [CrossRef]
- Prasad, A.; Shukla, S.P.; Mathur, A.; Chanotiya, C.S.; Mathur, A.K. Genetic fidelity of long-term micropropagated Lavandula officinalis Chaix.: An important aromatic medicinal plant. Plant Cell Tissue Organ Cult. PCTOC 2014, 120, 803–811. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; Salama, A.M.; Abou El-Leel, O.F. Analysis of genetic diversity of Lavandula species using taxonomic, essential oil and molecular genetic markers. Sciences 2017, 7, 141–154. [Google Scholar]
- Zagorcheva, T.; Stanev, S.; Rusanov, K.; Atanassov, I. SRAP markers for genetic diversity assessment of lavender (Lavandula angustifolia mill.) varieties and breeding lines. Biotechnol. Biotechnol. Equip. 2020, 34, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Adal, A.M.; Demissie, Z.A.; Mahmoud, S.S. Identification, validation and cross-species transferability of novel Lavandula EST-SSRs. Planta 2015, 241, 987–1004. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Alamer, K.H. Discriminating Lamiaceae Species from Saudi Arabia Using Allozyme and Specific DNA Markers. Pak. J. Bot. 2018, 50, 969–975. [Google Scholar]
- Adal, A.M. Development of Molecular Markers and Cloning of Genes Involved in The Biosynthesis of Monoterpenes in Lavandula; University of British Columbia: Vancouver, BC, Canada, 2019. [Google Scholar]
- Angioni, A.; Barra, A.; Coroneo, V.; Dessi, S.; Cabras, P. Chemical composition, seasonal variability, and antifungal activity of Lavandula stoechas L. ssp. stoechas essential oils from stem/leaves and flowers. J. Agric. Food Chem. 2006, 54, 4364–4370. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, T.; Dugo, G.; Ruberto, G.; Leto, C.; Napoli, E.M.; Potortì, A.G.; Fede, M.R.; Virga, G.; Leone, R.; Anna, E.D.; et al. Agronomical evaluation of Sicilian biotypes of Lavandula stoechas L. spp. stoechasand analysis of the essential oils. J. Essent. Oil Res. 2015, 27, 115–124. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Dong, Y.; Zhang, W.; Wang, D.; Bai, H.; Li, K.; Li, H.; Shi, L. The chromosome-based lavender genome provides new insights into Lamiaceae evolution and terpenoid biosynthesis. Hortic. Res. 2021, 8, 1–14. [Google Scholar]
- Stevanato, P.; Broccanello, C.; Biscarini, F.; Del Corvo, M.; Sablok, G.; Panella, L.; Stella, A.; Concheri, G. High-Throughput RAD-SNP Genotyping for Characterization of Sugar Beet Genotypes. Plant Mol. Biol. Report. 2013, 32, 691–696. [Google Scholar] [CrossRef]
- Rochette, N.C.; Rivera-Colon, A.G.; Catchen, J.M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef]
- Rohlf, F. NTSYS-pc: Numerical Taxonomy Multivariate Analysis System; Applied Biostatistics, I. & Exeter Software (Firm): Setauket, NY, USA, 2009. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Kakuta, M.; Ishida, T.; Akiyama, Y. GHOSTX: An improved sequence homology search algorithm using a query suffix array and a database suffix array. PLoS ONE 2014, 9, e103833. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolè, S.; Erickson, D.L.; Ambrosi, D.; Bellucci, E.; Lucchin, M.; Papa, R.; Kress, W.J.; Barcaccia, G. Biodiversity studies in Phaseolus species by DNA barcoding. Genome 2011, 54, 529–545. [Google Scholar] [CrossRef]
- Wojciechowski, M.F.; Lavin, M.; Sanderson, M. A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am. J. Bot. 2004, 91, 1846–1862. [Google Scholar] [CrossRef]
- Sang, T.; Crawford, D.; Stuessy, T. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 1997, 84, 1120. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.A.; Simpson, B.B. Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Syst. Bot. 2003, 28, 723–737. [Google Scholar]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Wang, J.; Wang, Z.; Du, X.; Yang, H.; Han, F.; Han, Y.; Yuan, F.; Zhang, L.; Peng, S.; Guo, E. A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L.) P. Beauv.] using RAD-seq. PLoS ONE 2017, 12, e0179717. [Google Scholar] [CrossRef]
- Zhang, F.; Kang, J.; Long, R.; Yu, L.X.; Wang, Z.; Zhao, Z.; Zhang, T.; Yang, Q. High-density linkage map construction and mapping QTL for yield and yield components in autotetraploid alfalfa using RAD-seq. BMC Plant Biol. 2019, 19, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Conteh, B.; Fang, L.; Xia, Q.; Nian, H. QTL mapping for soybean (Glycine max L.) leaf chlorophyll-content traits in a genotyped RIL population by using RAD-seq based high-density linkage map. BMC Genom. 2020, 21, 739. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Liu, H.; Yang, M.; Tao, Y.; Ma, H.; Wu, W.; Zuo, Y.; Zhao, Y. High-density genetic map construction and QTLs analysis of grain yield-related traits in sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 2014, 14, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Hu, Y.; Mao, B.; Xiang, H.; Shao, Y.; Pan, Y.; Sheng, X.; Li, Y.; Ni, X.; Xia, Y.; et al. Genetic analysis for rice grain quality traits in the YVB stable variant line using RAD-seq. Mol. Genet Genom. 2016, 291, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tao, Y.; Zheng, Z.; Shao, D.; Li, Z.; Sweetingham, M.W.; Buirchell, B.J.; Li, C. Rapid development of molecular markers by next-generation sequencing linked to a gene conferring phomopsis stem blight disease resistance for marker-assisted selection in lupin (Lupinus angustifolius L.) breeding. Theor. Appl. Genet 2013, 126, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Zong, J.; Luo, Z.; Chen, M.; Zhao, X.; Zhang, D.; Qi, Y.; Yuan, Z. Development of a RAD-Seq Based DNA Polymorphism Identification Software, AgroMarker Finder, and Its Application in Rice Marker-Assisted Breeding. PLoS ONE 2016, 11, e0147187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, H.; Uchida, T.; Tanaka, Y.; Katai, H.; Nagano, A.J.; Morita, A.; Ikka, T. Genomic predictions and genome-wide association studies based on RAD-seq of quality-related metabolites for the genomics-assisted breeding of tea plants. Sci. Rep. 2020, 10, 17480. [Google Scholar] [CrossRef]
- Kawamura, K.; Kawanabe, T.; Shimizu, M.; Nagano, A.J.; Saeki, N.; Okazaki, K.; Kaji, M.; Dennis, E.S.; Osabe, K.; Fujimoto, R. Genetic distance of inbred lines of Chinese cabbage and its relationship to heterosis. Plant Gene 2016, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Jin, X.; Chen, N.; Li, X.; Li, P.; Fu, C. Phylogeny of Morella rubra and Its Relatives (Myricaceae) and Genetic Resources of Chinese Bayberry Using RAD Sequencing. PLoS ONE 2015, 10, e0139840. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Zhao, S.; Li, M.; Zhang, C.; Qu, H.; Li, Q.; Li, J.; Lin, Y.; Pu, Z. Genome-wide genetic diversity detection and population structure analysis in sweetpotato (Ipomoea batatas) using RAD-seq. Genomics 2020, 112, 1978–1987. [Google Scholar] [CrossRef]
- Moja, S.; Guitton, Y.; Nicolè, F.; Legendre, L.; Pasquier, B.; Upson, T.; Jullien, F. Genome size and plastid trnK-matK markers give new insights into the evolutionary history of the genus Lavandula L. Plant Biosyst. Int. J. Deal. All Asp. Plant Biol. 2015, 150, 1216–1224. [Google Scholar] [CrossRef]
- Robb, J.B.; Cherry, R.J. Multi-Bracted Lavender Plants. U.S. Patent No. 9301463, 5 April 2016. [Google Scholar]
- Lam, H.Y.; Clark, M.J.; Chen, R.; Chen, R.; Natsoulis, G.; O’huallachain, M.; Dewey, F.E.; Habegger, L.; Ashley, E.A.; Gerstein, M.B. Performance comparison of whole-genome sequencing platforms. Nat. Biotechnol. 2012, 30, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eklöf, H.; Bernhardsson, C.; Ingvarsson, P.K. Comparing the Effectiveness of Exome Capture Probes, Genotyping by Sequencing and Whole-Genome Re-Sequencing for Assessing Genetic Diversity in Natural and Managed Stands of Picea abies. Forests 2020, 11, 1185. [Google Scholar] [CrossRef]
- Rabbi, I.Y.; Kulakow, P.A.; Manu-Aduening, J.A.; Dankyi, A.A.; Asibuo, J.Y.; Parkes, E.Y.; Abdoulaye, T.; Girma, G.; Gedil, M.A.; Ramu, P.; et al. Tracking crop varieties using genotyping-by-sequencing markers: A case study using cassava (Manihot esculenta Crantz). BMC Genet 2015, 16, 115. [Google Scholar] [CrossRef] [Green Version]
- McCoy, R.M.; Utturkar, S.M.; Crook, J.W.; Thimmapuram, J.; Widhalm, J.R. The origin and biosynthesis of the naphthalenoid moiety of juglone in black walnut. Hortic. Res. 2018, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, M.; Xiong, Y.; Yan, H.; Zhang, X.; Li, Y.; da Silva, J.A.T.; Ma, G. Cloning and Expression Analysis of Mevalonate Kinase and Phosphomevalonate Kinase Genes Associated with MVA Pathway in Santalum Album. Sci. Rep. 2020, 11, 1–13. [Google Scholar]
- Achard, F.; Butruille, M.; Madjarac, S.; Nelson, P.; Duesing, J.; Laffont, J.L.; Nelson, B.; Xiong, J.; Mikel, M.A.; Smith, J. Single nucleotide polymorphisms facilitate distinctness-uniformity-stability testing of soybean cultivars for plant variety protection. Crop Sci. 2020, 60, 2280–2303. [Google Scholar] [CrossRef]
- Jamali, S.H.; Cockram, J.; Hickey, L.T. Insights into deployment of DNA markers in plant variety protection and registration. Theor. Appl. Genet 2019, 132, 1911–1929. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-K.; Chung, Y.-S. Plant Variety Protection: Current Practices and Insights. Genes 2021, 12, 1127. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Marker | Primer Name | Primer Sequence (5′-3′) * | Ta (°C) | References |
|---|---|---|---|---|
| rbcL gene (cpDNA) | rbcL_F | GCAGCATTYCGAGTAASTCCYCA | 55 | [30] |
| rbcL_R | GAAACGYTCTCTCCAWCGCATAAA | [30] | ||
| matK gene (cpDNA) | matK4La | CCTTCGATACTGGGTGAAAGAT | 55 | [31] |
| matK1932Ra | CCAGACCGGCTTACTAATGGG | [31] | ||
| trnH-psbA (cpDNA) | psbA3′f | GTTATGCATGAACGTAATGCTC | 55 | [32] |
| trnHf | CGCATGGTGGATTCACAATCC | [33] | ||
| ITS1 (nuDNA) | ITS5 | GGAAGTAAAAGTCGTAACAAGG | 55 | [34] |
| ITS2 | GCTGCGTTCTTCATCGATGC | [34] |
| Obs. Ho | Obs. He | Sample | Genetic Similarity (GS) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1603 | 66.1% | 33.9% | Cluster A | 1603 | 100.0% | ||||||||||||||
| 2601 | 60.1% | 39.9% | 2601 | 82.8% | 100.0% | ||||||||||||||
| 2604 | 72.8% | 27.2% | 2604 | 78.9% | 77.6% | 100.0% | |||||||||||||
| 1605 | 76.4% | 23.6% | Cluster B | 1605 | 79.8% | 77.8% | 73.8% | 100.0% | |||||||||||
| 1841 | 78.8% | 21.2% | 1841 | 77.9% | 75.7% | 71.1% | 86.4% | 100.0% | |||||||||||
| 1826 | 77.9% | 22.1% | 1826 | 79.2% | 76.9% | 74.1% | 87.4% | 86.5% | 100.0% | ||||||||||
| SD-014 | 85.5% | 14.5% | SD-014 | 76.5% | 74.3% | 70.6% | 88.5% | 83.9% | 83.3% | 100.0% | |||||||||
| BPI | 90.1% | 9.9% | Cluster C | BPI | 74.6% | 72.3% | 68.2% | 82.8% | 79.1% | 83.7% | 83.8% | 100.0% | |||||||
| ST-913 | 84.8% | 15.2% | ST-913 | 75.0% | 74.2% | 70.4% | 85.7% | 81.1% | 86.5% | 83.5% | 93.3% | 100.0% | |||||||
| SD-332 | 82.4% | 17.6% | SD-332 | 75.9% | 74.4% | 70.5% | 83.5% | 79.1% | 84.8% | 85.4% | 93.7% | 93.5% | 100.0% | ||||||
| 1811 | 89.7% | 10.3% | 1811 | 75.1% | 72.1% | 67.7% | 86.0% | 85.6% | 85.9% | 86.4% | 89.7% | 92.2% | 89.5% | 100.0% | |||||
| ST-103 | 77.6% | 22.4% | Cluster D | ST-103 | 75.7% | 72.9% | 69.4% | 80.7% | 79.4% | 76.9% | 82.8% | 83.2% | 82.2% | 82.3% | 82.0% | 100.0% | |||
| 3601 | 78.1% | 21.9% | 3601 | 72.2% | 70.4% | 67.8% | 78.0% | 76.5% | 75.2% | 80.0% | 76.3% | 77.2% | 77.2% | 80.9% | 86.0% | 100.0% | |||
| 2603 | 87.8% | 12.2% | Cluster E | 2603 | 63.0% | 64.0% | 65.9% | 55.4% | 58.8% | 56.2% | 54.0% | 53.9% | 53.9% | 53.4% | 51.6% | 54.9% | 53.1% | 100.0% | |
| 2605 | 71.6% | 28.4% | 2605 | 67.6% | 68.9% | 69.3% | 62.0% | 66.4% | 62.9% | 60.0% | 58.9% | 61.0% | 60.3% | 59.7% | 64.0% | 63.8% | 73.7% | 100.0% | |
| 1603 | 2601 | 2604 | 1605 | 1841 | 1826 | SD-014 | BPI | ST-913 | SD-332 | 1811 | ST-103 | 3601 | 2603 | 2605 | |||||
| Cluster A | Cluster B | Cluster C | Cluster D | Cluster E | |||||||||||||||
| Avg. Obs. Ho | Cluster | Avg. Genetic Similarity (GS) | |||||
|---|---|---|---|---|---|---|---|
| 66.4% ± 3.7% | Cluster A | 79.8% ± 1.6% | |||||
| 79.7% ± 2.0% | Cluster B | 75.6% ± 0.9% | 86.0% ± 0.8% | ||||
| 86.7% ± 1.9% | Cluster C | 72.5% ± 0.8% | 83.9% ± 0.6% | 92.0% ± 0.8% | |||
| 77.9% ± 0.2% | Cluster D | 71.4% ± 1.1% | 78.7% ± 0.9% | 80.2% ± 1.0% | 86.0% ± N/A | ||
| 79.7% ± 8.1% | Cluster E | 66.4% ± 1.1% | 59.4% ± 1.5% | 56.6% ± 1.3% | 58.9% ± 2.9% | 73.7% ± N/A | |
| 78.5% ± 2.4% | A + B + C + D | 60.1% ± 1.0% | 79.7% ± 0.7% | ||||
| Cluster A | Cluster B | Cluster C | Cluster D | Cluster E | A + B + C + D | ||
| BLASTn Result | RAD-Tags (n) | CDS (n) | Protein Products (n) | Avg. Identity (%) | Avg. Length (bp) | Avg. E-Value | Avg. Bitscore | Avg. Score | Avg. Mismatches (n) | Avg. Identity (n) | Avg. Positive Positions |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exome S.ind | 2618 | 2907 | 2077 | 87.3 | 64.4 | 5.33 × 10−12 | 80.2 | 87.5 | 8.2 | 56.2 | 87.3 |
| Flavonoids | 15 | 14 | 10 | 86.7 | 67.1 | 1.04 × 10−12 | 82.1 | 89.6 | 8.9 | 58.2 | 86.7 |
| Terpenes | 20 | 24 | 19 | 86.0 | 62.9 | 6.20 × 10−12 | 74.3 | 81.0 | 9.0 | 53.9 | 86.0 |
| Exome S.sp | 4239 | 6534 | 1215 | 88.7 | 64.2 | 2.90 × 10−12 | 83.8 | 91.5 | 7.3 | 56.9 | 88.7 |
| Flavonoids | 33 | 40 | 18 | 87.4 | 66.0 | 2.41 × 10−12 | 82.5 | 90.1 | 8.3 | 57.6 | 87.4 |
| Terpenes | 61 | 65 | 28 | 88.9 | 65.6 | 1.45 × 10−12 | 86.6 | 94.7 | 7.3 | 58.3 | 88.9 |
| Obs. Ho | Obs. He | Genetic Similarity (GS) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 68.3% | 31.7% | Cluster A | 1603 | 100.0% | ||||||||||||||
| 60.4% | 39.6% | 2601 | 83.1% | 100.0% | ||||||||||||||
| 73.7% | 26.3% | 2604 | 79.6% | 77.3% | 100.0% | |||||||||||||
| 78.3% | 21.7% | Cluster B | 1605 | 81.2% | 78.5% | 74.8% | 100.0% | |||||||||||
| 86.1% | 13.9% | SD-014 | 77.9% | 75.5% | 72.3% | 88.9% | 100.0% | |||||||||||
| 78.2% | 21.8% | 1841 | 79.7% | 77.5% | 73.1% | 86.9% | 85.1% | 100.0% | ||||||||||
| 79.5% | 20.5% | 1826 | 81.0% | 78.3% | 75.9% | 87.0% | 84.5% | 88.1% | 100.0% | |||||||||
| 90.4% | 9.6% | Cluster C | BPI | 76.4% | 74.0% | 70.6% | 83.5% | 85.8% | 80.6% | 84.3% | 100.0% | |||||||
| 85.3% | 14.7% | ST-913 | 76.9% | 76.1% | 72.7% | 86.2% | 85.5% | 82.0% | 87.4% | 94.2% | 100.0% | |||||||
| 83.2% | 16.8% | SD-332 | 77.3% | 76.3% | 72.9% | 83.9% | 86.9% | 80.4% | 85.1% | 93.9% | 93.7% | 100.0% | ||||||
| 89.7% | 10.3% | 1811 | 77.5% | 74.3% | 70.4% | 86.8% | 87.9% | 86.2% | 86.6% | 90.4% | 92.6% | 90.0% | 100.0% | |||||
| 78.5% | 21.5% | Cluster D | ST-103 | 77.5% | 74.7% | 71.3% | 82.5% | 84.2% | 81.6% | 79.0% | 84.0% | 83.8% | 83.3% | 84.1% | 100.0% | |||
| 77.3% | 22.7% | 3601 | 75.0% | 73.2% | 70.4% | 79.4% | 80.9% | 78.5% | 76.6% | 77.8% | 78.9% | 78.4% | 82.2% | 87.0% | 100.0% | |||
| 87.0% | 13.0% | Cluster E | 2603 | 65.8% | 67.4% | 68.9% | 59.0% | 58.9% | 63.0% | 61.1% | 57.9% | 58.2% | 58.1% | 56.4% | 58.7% | 57.8% | 100.0% | |
| 70.2% | 29.8% | 2605 | 69.1% | 70.4% | 70.9% | 64.1% | 63.5% | 69.2% | 65.9% | 61.5% | 63.6% | 63.3% | 62.9% | 66.5% | 67.4% | 74.3% | 100.0% | |
| 1603 | 2601 | 2604 | 1605 | SD-014 | 1841 | 1826 | BPI | ST-913 | SD-332 | 1811 | ST-103 | 3601 | 2603 | 2605 | ||||
| Cluster A | Cluster B | Cluster C | Cluster D | Cluster E | ||||||||||||||
| Avg. Obs. Ho | Sample | Avg. Genetic Similarity (GS) | |||||
|---|---|---|---|---|---|---|---|
| 67.4% ± 3.9% | Cluster A | 80.0% ± 1.7% | |||||
| 80.5% ± 1.9% | Cluster B | 77.1% ± 0.8% | 86.7% ± 0.7% | ||||
| 87.2% ± 1.7% | Cluster C | 74.6% ± 0.7% | 84.9% ± 0.6% | 92.5% ± 0.8% | |||
| 77.9% ± 0.4% | Cluster D | 73.7% ± 1.1% | 80.3% ± 0.9% | 81.6% ± 1.0% | 87.0% ± N/A | ||
| 78.6% ± 8.4% | Cluster E | 68.8% ± 0.8% | 63.1% ± 1.2% | 60.2% ± 1.0% | 62.6% ± 2.5% | 74.3% ± N/A | |
| 79.1% ± 2.3% | A + B + C + D | 63.4% ± 0.9% | 81.0% ± 0.7% | ||||
| Cluster A | Cluster B | Cluster C | Cluster D | Cluster E | A + B + C + D | ||
| FLAVONOIDS | ||||
|---|---|---|---|---|
| Read ID | S. ind CDS ID | product | KO-IDs from KEGG | SNP to AA Subs. |
| 3043 | XP_011100449.1 | anthocyanidin 3-O-glucosyltransferase 2 | K12930 | Ile -> Met |
| XP_011100453.1 | anthocyanidin 3-O-glucosyltransferase 2-like | |||
| 6706 | XP_011090466.1 | aspartate aminotransferase and glu/asp-prephenate aminotransferase | K15849 | Val -> Ala |
| 7480 | XP_011089364.1 | arogenate dehydratase/prephenate dehydratase 2, chloroplastic | K05359 | Glu -> Val |
| XP_011089363.1 | ||||
| 7969 | XP_011094662.1 | phenylalanine ammonia-lyase | K10775 | Gln -> Arg |
| 9011 | XP_011089239.2 | LOW QUALITY PROTEIN: 4-coumarate--CoA ligase-like 7 | K01904 | Gln -> Gln |
| 9012 | Gln -> Arg | |||
| 9955 | XP_020554052.1 | putative anthocyanidin reductase isoform X2 | K08695 | Uncertain |
| XP_011095308.1 | X -> Leu | |||
| 10947 | XP_011069886.1 | anthocyanidin 3-O-glucosyltransferase-like | K12930 | Arg -> Pro |
| 11587 | XP_011077338.1 | phenylalanine ammonia-lyase | K10775 | His -> Tyr |
| TERPENES | ||||
| Read ID | S. ind CDS_ID | product | KO-IDs from KEGG | SNP to AA Subs. |
| 8036 | XP_011071094.1 | 1,4-dihydroxy-2-naphthoyl-CoA synthase, peroxisomal | K01661 | X -> X |
| 14576 | XP_011096130.1 | α-farnesene synthase | K14173 | Gly -> Glu |
| 6208 | XP_011093795.1 | β-amyrin synthase | K15813 | Lys -> Glu |
| 8386 | XP_011093795.1 | X -> Arg | ||
| 6208 | XP_011085901.1 | β-amyrin synthase-like | K15813 | Lys -> Glu |
| 8386 | XP_011085901.1 | X -> Arg | ||
| 6276 | XP_011095756.1 | ent-kaur-16-ene synthase, chloroplastic | N/A | Pro -> Ala |
| 7199 | XP_011083784.1 | ent-kaurene oxidase, chloroplastic-like | K04122 | Val -> Met |
| 3576 | XP_020550121.1 | geranylgeranyl transferase type-2 subunit α 1 | K09833 | Leu -> Ser |
| 10802 | XP_011092247.1 | gibberellin 20-oxidase-like protein | K05282 | Gln -> Gln |
| 11279 | XP_011096560.1 | gibberellin 2-β-dioxygenase | K04125 | Phe -> Leu |
| 10014 | XP_011098626.1 | gibberellin-regulated protein 4-like | N/A | Arg -> Gln |
| XP_011071640.1 | ||||
| 4578 | XP_011084658.1 | isopentenyl-diphosphate Delta-isomerase I | K01823 | Uncertain |
| 6515 | Phe -> Leu | |||
| 13525 | XP_011077171.1 | Pro -> Pro | ||
| 9817 | XP_011075409.1 | probable NAD(P)H dehydrogenase subunit CRR3, chloroplastic | N/A | Trp -> Leu |
| 14513 | XP_011082816.1 | probable solanesyl-diphosphate synthase 3, chloroplastic | K05356 | Leu -> Phe |
| 14513 | XP_011098150.1 | probable solanesyl-diphosphate synthase 3, chloroplastic isoform X2 | Leu -> Phe | |
| 5640 | XP_020551000.1 | protein prenyltransferase α subunit, isoform X6 | K14137 | Pro -> Gln |
| XP_020551002.1 | ||||
| 3603 | XP_011078470.1 | squalene monooxygenase | K00511 | Asn -> Thr |
| 9296 | XP_011092466.1 | squalene monooxygenase-like | Asp -> His | |
| 5280 | XP_011092839.1 | squalene synthase | K00801 | Pro -> Ser |
| XP_011092841.1 | ||||
| 4990 | XP_011082248.1 | vetispiradiene synthase 3 isoform X2 | K14182 | Asp -> Glu |
| 14152 | XP_020548233.1 | isochorismate synthase, chloroplastic-like | K01851 | Arg -> Met |
| 14154 | Gln -> Pro | |||
| 14685 | XP_020548234.1 | isochorismate synthase, chloroplastic-like | K01851 | Val -> Leu |
| 14687 | Thr -> Thr | |||
| 15015 | Lys -> Lys |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scariolo, F.; Palumbo, F.; Vannozzi, A.; Sacilotto, G.B.; Gazzola, M.; Barcaccia, G. Genotyping Analysis by RAD-Seq Reads Is Useful to Assess the Genetic Identity and Relationships of Breeding Lines in Lavender Species Aimed at Managing Plant Variety Protection. Genes 2021, 12, 1656. https://doi.org/10.3390/genes12111656
Scariolo F, Palumbo F, Vannozzi A, Sacilotto GB, Gazzola M, Barcaccia G. Genotyping Analysis by RAD-Seq Reads Is Useful to Assess the Genetic Identity and Relationships of Breeding Lines in Lavender Species Aimed at Managing Plant Variety Protection. Genes. 2021; 12(11):1656. https://doi.org/10.3390/genes12111656
Chicago/Turabian StyleScariolo, Francesco, Fabio Palumbo, Alessandro Vannozzi, Gio Batta Sacilotto, Marco Gazzola, and Gianni Barcaccia. 2021. "Genotyping Analysis by RAD-Seq Reads Is Useful to Assess the Genetic Identity and Relationships of Breeding Lines in Lavender Species Aimed at Managing Plant Variety Protection" Genes 12, no. 11: 1656. https://doi.org/10.3390/genes12111656
APA StyleScariolo, F., Palumbo, F., Vannozzi, A., Sacilotto, G. B., Gazzola, M., & Barcaccia, G. (2021). Genotyping Analysis by RAD-Seq Reads Is Useful to Assess the Genetic Identity and Relationships of Breeding Lines in Lavender Species Aimed at Managing Plant Variety Protection. Genes, 12(11), 1656. https://doi.org/10.3390/genes12111656