
Signaling Pathways That Regulate Normal and Aberrant Red Blood Cell Development


{kind=link}
{kind=link}
Abstract
1. Introduction
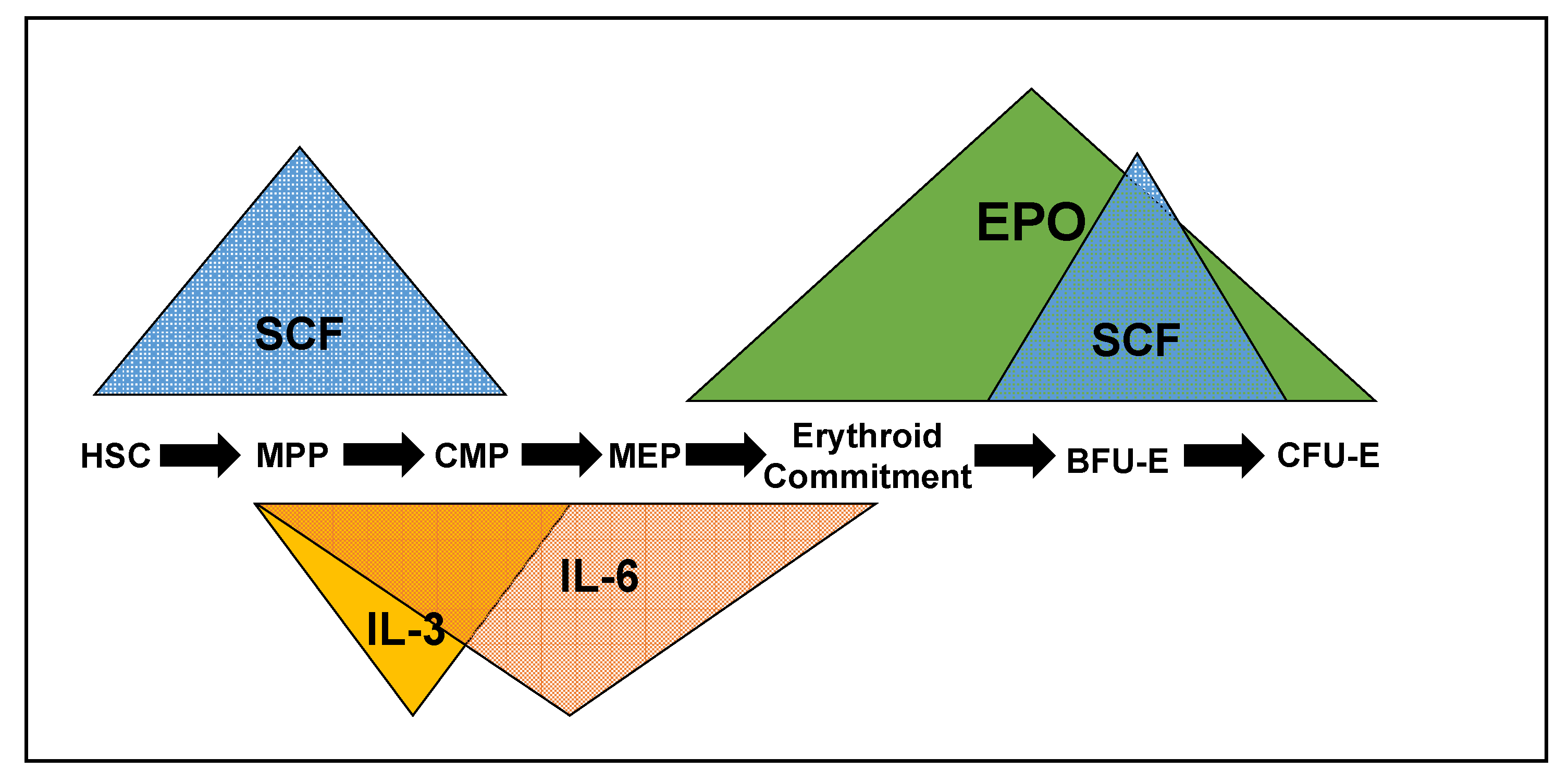
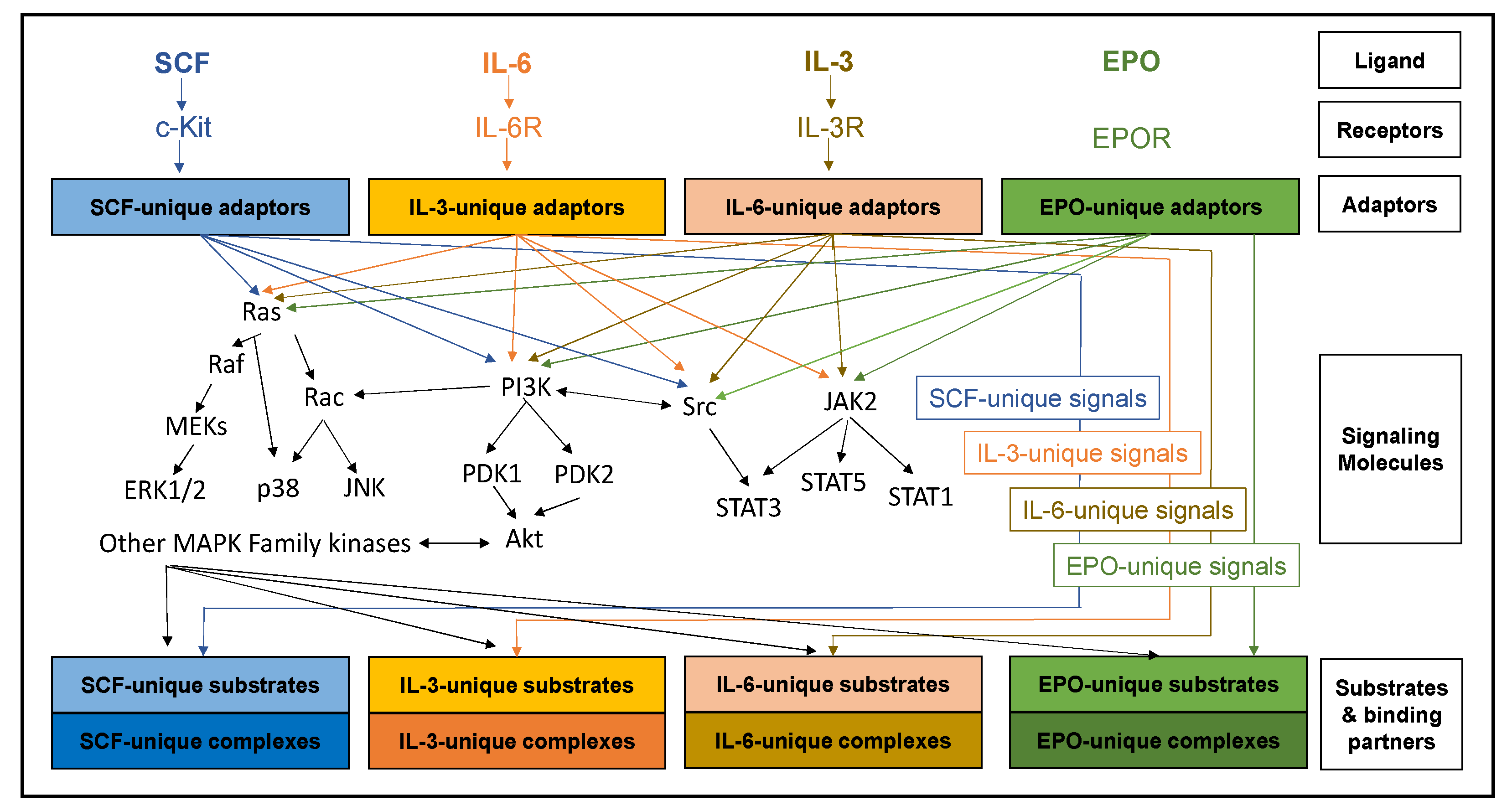
2. Cytokines, Cognate Receptors and Signaling Pathways Regulating Early Erythropoiesis
3. Other Cytokines and Signaling Pathways
4. Kinases and Anemia
5. Summary
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dygai, A.M.; Zhdanov, V.V.; Miroshnichenko, L.A.; Zyuz’kov, G.N.; Udut, E.V.; Simanina, E.V.; Stavrova, L.A.; Khrichkova, T.Y.; Reykhart, D.V.; Agafonov, V.I. Mechanisms of stimulating effect of glycyram and D-glucuronic acid on granulocytopoiesis suppression by 5-fluorouracil. Bull. Exp. Biol. Med. 2013, 155, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Dygai, A.M.; Zhdanov, V.V.; Miroshnichenko, L.A.; Udut, E.V.; Zyuz’kov, G.N.; Khrichkova, T.Y.; Simanina, E.V.; Sherstoboev, E.Y.; Stavrova, L.A.; Chaikovskii, A.V.; et al. Role of PI3K, ERK, and p38 Signaling Pathways in the Production of Humoral Erythropoiesis Regulators under Normal Conditions. Bull. Exp. Biol. Med. 2015, 160, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Dygai, A.M.; Zhdanov, V.V.; Miroshnichenko, L.A.; Udut, E.V.; Zyuz’kov, G.N.; Simanina, E.V.; Chaikovskii, A.V.; Stavrova, L.A.; Trofimova, E.S.; Burmina, Y.V. Participation of signaling cascades in the regulation of erythropoiesis under conditions of cytostatic treatment. Bull. Exp. Biol. Med. 2015, 158, 304–307. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef]
- Alexander, W.S. Cytokines in hematopoiesis. Int. Rev. Immunol. 1998, 16, 651–682. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, R.; Joachimiak, A.; Schlessinger, J.; Kong, X.P. Crystal structure of human stem cell factor: Implication for stem cell factor receptor dimerization and activation. Proc. Natl. Acad. Sci. USA 2000, 97, 7732–7737. [Google Scholar] [CrossRef]
- Oriss, T.B.; Krishnamoorthy, N.; Ray, P.; Ray, A. Dendritic cell c-kit signaling and adaptive immunity: Implications for the upper airways. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 7–12. [Google Scholar] [CrossRef]
- Ogawa, M.; Nishikawa, S.; Yoshinaga, K.; Hayashi, S.; Kunisada, T.; Nakao, J.; Kina, T.; Sudo, T.; Kodama, H.; Nishikawa, S. Expression and function of c-Kit in fetal hemopoietic progenitor cells: Transition from the early c-Kit-independent to the late c-Kit-dependent wave of hemopoiesis in the murine embryo. Development 1993, 117, 1089–1098. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Opatowsky, Y.; Zhang, Z.; Mandiyan, V.; Lax, I.; Schlessinger, J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 2007, 130, 323–334. [Google Scholar] [CrossRef]
- Lev, S.; Givol, D.; Yarden, Y. Interkinase domain of kit contains the binding site for phosphatidylinositol 3’ kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 678–682. [Google Scholar] [CrossRef]
- Ueki, K.; Fruman, D.A.; Brachmann, S.M.; Tseng, Y.H.; Cantley, L.C.; Kahn, C.R. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol. Cell. Biol. 2002, 22, 965–977. [Google Scholar] [CrossRef]
- Nair, A.; Chakraborty, S.; Banerji, L.A.; Srivastava, A.; Navare, C.; Saha, B. Ras isoforms: Signaling specificities in CD40 pathway. Cell Commun. Signal. 2020, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. Does Ras Activate Raf and PI3K Allosterically? Front. Oncol. 2019, 9, 1231. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Kaibori, Y.; Saito, Y.; Nakayama, Y. EphA2 phosphorylation at Ser897 by the Cdk1/MEK/ERK/RSK pathway regulates M-phase progression via maintenance of cortical rigidity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 5334–5349. [Google Scholar] [CrossRef] [PubMed]
- Badhai, J.; Fröjmark, A.S.; Davey, E.J.; Schuster, J.; Dahl, N. Ribosomal protein S19 and S24 insufficiency cause distinct cell cycle defects in Diamond-Blackfan anemia. Biochim. Biophys. Acta 2009, 1792, 1036–1042. [Google Scholar] [CrossRef]
- Noy-Lotan, S.; Dgany, O.; Lahmi, R.; Marcoux, N.; Krasnov, T.; Yissachar, N.; Ginsberg, D.; Motro, B.; Resnitzky, P.; Yaniv, I.; et al. Codanin-1, the protein encoded by the gene mutated in congenital dyserythropoietic anemia type I (CDAN1), is cell cycle-regulated. Haematologica 2009, 94, 629–637. [Google Scholar] [CrossRef]
- Ashley, R.J.; Yan, H.; Wang, N.; Hale, J.; Dulmovits, B.M.; Papoin, J.; Olive, M.E.; Udeshi, N.D.; Carr, S.A.; Vlachos, A.; et al. Steroid-resistance in Diamond Blackfan anemia associates with p57Kip2 dysregulation in erythroid progenitors. J. Clin. Investig. 2020, 130, 2097–2110. [Google Scholar] [CrossRef]
- Grace, R.F.; Zanella, A.; Neufeld, E.J.; Morton, D.H.; Eber, S.; Yaish, H.; Glader, B. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am. J. Hematol. 2015, 90, 825–830. [Google Scholar] [CrossRef]
- Wilkes, M.C.; Siva, K.; Chen, J.; Varetti, G.; Youn, M.Y.; Chae, H.; Ek, F.; Olsson, R.; Lundbäck, T.; Dever, D.P.; et al. Diamond Blackfan anemia is mediated by hyperactive Nemo-like kinase. Nat. Commun. 2020, 11, 3344. [Google Scholar] [CrossRef]
- Wilkes, M.C.; Siva, K.; Varetti, G.; Mercado, J.; Wentworth, E.P.; Perez, C.; Saxena, M.; Kam, S.; Kapur, S.; Chen, J.; et al. Metformin-induced suppression of NLK improves erythropoiesis in Diamond Blackfan Anemia through induction of miR-26a. Exp. Hematol. 2020, 91, 65–77. [Google Scholar] [CrossRef]
- Wilkes, M.C.; Jung, K.; Lee, B.E.; Saxena, M.; Sathianathen, R.S.; Mercado, J.D.; Perez, C.; Flygare, J.; Narla, A.; Glader, B.; et al. The active component of Ginseng, Ginsenoside Rb1, improves erythropoiesis in models of Diamond Blackfan Anemia by targeting Nemo-like Kinase. J. Biol. Chem. 2021, 297, 100988. [Google Scholar] [CrossRef]
- Chartier, M.E.; Hart, L.; Paganelli, M.; Ahmed, N.; Bilodeau, M.; Alvarez, F. Successful Liver Transplants for Liver Failure Associated With Pyruvate Kinase Deficiency. Pediatrics 2018, 141, S385–S389. [Google Scholar] [CrossRef] [PubMed]
- Bogacheva, O.; Bogachev, O.; Menon, M.; Dev, A.; Houde, E.; Valoret, E.I.; Prosser, H.M.; Creasy, C.L.; Pickering, S.J.; Grau, E.; et al. DYRK3 dual-specificity kinase attenuates erythropoiesis during anemia. J. Biol. Chem. 2008, 283, 36665–36675. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tran, J.; Wang, H.; Guo, C.; Harro, D.; Campbell, A.D.; Eitzman, D.T. mTOR Inhibition improves anaemia and reduces organ damage in a murine model of sickle cell disease. Br. J. Haematol. 2016, 174, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Pearl-Yafe, M.; Halperin, D.; Scheuerman, O.; Fabian, I. The p38 pathway partially mediates caspase-3 activation induced by reactive oxygen species in Fanconi anemia C cells. Biochem. Pharm. 2004, 67, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Chopra, R.; Pu, Q.Q.; Elefanty, A.G. Biology of BCR-ABL. Blood Rev. 1999, 13, 211–229. [Google Scholar] [CrossRef]
- Gommerman, J.L.; Sittaro, D.; Klebasz, N.Z.; Williams, D.A.; Berger, S.A. Differential stimulation of c-Kit mutants by membrane-bound and soluble Steel Factor correlates with leukemic potential. Blood 2000, 96, 3734–3742. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Clark, S.C. Human interleukin 3: Analysis of the gene and its role in the regulation of hematopoiesis. Int. J. Cell Cloning 1990, 8 (Suppl. 1), 121–128. [Google Scholar] [CrossRef]
- Ihle, J.N.; Keller, J.; Oroszlan, S.; Henderson, L.E.; Copeland, T.D.; Fitch, F.; Prystowsky, M.B.; Goldwasser, E.; Schrader, J.W.; Palaszynski, E.; et al. Biologic properties of homogeneous interleukin 3. I. Demonstration of WEHI-3 growth factor activity, mast cell growth factor activity, p cell-stimulating factor activity, colony-stimulating factor activity, and histamine-producing cell-stimulating factor activity. J. Immunol. 1983, 131, 282–287. [Google Scholar]
- Metcalf, D. The molecular control of cell division, differentiation commitment and maturation in haemopoietic cells. Nature 1989, 339, 27–30. [Google Scholar] [CrossRef]
- Pierce, J.H. Oncogenes, growth factors and hematopoietic cell transformation. Biochim. Biophys. Acta 1989, 989, 179–208. [Google Scholar] [CrossRef]
- Reddy, E.P.; Korapati, A.; Chaturvedi, P.; Rane, S. IL-3 signaling and the role of Src kinases, JAKs and STATs: A covert liaison unveiled. Oncogene 2000, 19, 2532–2547. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Hirano, T. Association of p72 tyrosine kinase with Stat factors and its activation by interleukin-3, interleukin-6, and granulocyte colony-stimulating factor. Blood 1994, 83, 3457–3461. [Google Scholar] [CrossRef]
- Jaster, R.; Zhu, Y.; Pless, M.; Bhattacharya, S.; Mathey-Prevot, B.; D’Andrea, A.D. JAK2 is required for induction of the murine DUB-1 gene. Mol. Cell Biol. 1997, 17, 3364–3372. [Google Scholar] [CrossRef] [PubMed]
- Silvennoinen, O.; Witthuhn, B.A.; Quelle, F.W.; Cleveland, J.L.; Yi, T.; Ihle, J.N. Structure of the murine Jak2 protein-tyrosine kinase and its role in interleukin 3 signal transduction. Proc. Natl. Acad. Sci. USA 1993, 90, 8429–8433. [Google Scholar] [CrossRef] [PubMed]
- Quelle, F.W.; Sato, N.; Witthuhn, B.A.; Inhorn, R.C.; Eder, M.; Miyajima, A.; Griffin, J.D.; Ihle, J.N. JAK2 associates with the beta c chain of the receptor for granulocyte-macrophage colony-stimulating factor, and its activation requires the membrane-proximal region. Mol. Cell Biol. 1994, 14, 4335–4341. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Nakafuku, M.; Miyajima, A.; Kaziro, Y. Involvement of ras p21 protein in signal-transduction pathways from interleukin 2, interleukin 3, and granulocyte/macrophage colony-stimulating factor, but not from interleukin 4. Proc. Natl. Acad. Sci. USA 1991, 88, 3314–3318. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Pazdrak, K.; Stafford, S.; Forsythe, P. The interleukin-5/receptor interaction activates Lyn and Jak2 tyrosine kinases and propagates signals via the Ras-Raf-1-MAP kinase and the Jak-STAT pathways in eosinophils. Int. Arch. Allergy Immunol. 1995, 107, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Coffer, P.J.; Geijsen, N.; M’Rabet, L.; Schweizer, R.C.; Maikoe, T.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L. Comparison of the roles of mitogen-activated protein kinase kinase and phosphatidylinositol 3-kinase signal transduction in neutrophil effector function. Biochem. J. 1998, 329 Pt 1, 121–130. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef]
- Malik, J.; Kim, A.R.; Tyre, K.A.; Cherukuri, A.R.; Palis, J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013, 98, 1778–1787. [Google Scholar] [CrossRef]
- Hirano, I.; Suzuki, N. The Neural Crest as the First Production Site of the Erythroid Growth Factor Erythropoietin. Front. Cell Dev. Biol. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; Koniski, A. Functional Analysis of Erythroid Progenitors by Colony-Forming Assays. Methods Mol. Biol. 2018, 1698, 117–132. [Google Scholar] [PubMed]
- Dame, C.; Fahnenstich, H.; Freitag, P.; Hofmann, D.; Abdul-Nour, T.; Bartmann, P.; Fandrey, J. Erythropoietin mRNA expression in human fetal and neonatal tissue. Blood 1998, 92, 3218–3225. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Rajvanshi, P.K.; Noguchi, C.T. The Many Facets of Erythropoietin Physiologic and Metabolic Response. Front. Physiol. 2019, 10, 1534. [Google Scholar] [CrossRef] [PubMed]
- Recny, M.A.; Scoble, H.A.; Kim, Y. Structural characterization of natural human urinary and recombinant DNA-derived erythropoietin. Identification of des-arginine 166 erythropoietin. J. Biol. Chem. 1987, 262, 17156–17163. [Google Scholar] [CrossRef]
- Liongue, C.; Sertori, R.; Ward, A.C. Evolution of Cytokine Receptor Signaling. J. Immunol. 2016, 197, 11–18. [Google Scholar] [CrossRef]
- De Marchi, S.; Pirisi, M.; Ferraccioli, G.F. Erythropoietin and the anemia of chronic diseases. Clin. Exp. Rheumatol. 1993, 11, 429–444. [Google Scholar]
- Narla, A.; Vlachos, A.; Nathan, D.G. Diamond Blackfan anemia treatment: Past, present, and future. Semin. Hematol. 2011, 48, 117–123. [Google Scholar] [CrossRef]
- Vlachos, A.; Atsidaftos, E.; Lababidi, M.L.; Muir, E.; Rogers, Z.R.; Alhushki, W.; Bernstein, J.; Glader, B.; Gruner, B.; Hartung, H.; et al. L-leucine improves anemia and growth in patients with transfusion-dependent Diamond-Blackfan anemia: Results from a multicenter pilot phase I/II study from the Diamond-Blackfan Anemia Registry. Pediatr. Blood Cancer 2020, 67, e28748. [Google Scholar] [CrossRef]
- Fulzele, K.; Krause, D.S.; Panaroni, C.; Saini, V.; Barry, K.J.; Liu, X.; Lotinun, S.; Baron, R.; Bonewald, L.; Feng, J.Q.; et al. Myelopoiesis is regulated by osteocytes through Gsα-dependent signaling. Blood 2013, 121, 930–939. [Google Scholar] [CrossRef]
- Kumar, A.; Jaggi, A.S.; Singh, N. Pharmacology of Src family kinases and therapeutic implications of their modulators. Fundam. Clin. Pharm. 2015, 29, 115–130. [Google Scholar] [CrossRef]
- Lennartsson, J.; Blume-Jensen, P.; Hermanson, M.; Pontén, E.; Carlberg, M.; Rönnstrand, L. Phosphorylation of Shc by Src family kinases is necessary for stem cell factor receptor/c-kit mediated activation of the Ras/MAP kinase pathway and c-fos induction. Oncogene 1999, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Monaco, G.; Sun, T.; Liu, J.; Lin, H.; Stephens, C.; Belmont, J.; Arlinghaus, R.B. BCR gene expression blocks Bcr-Abl induced pathogenicity in a mouse model. Oncogene 2001, 20, 1873–1881. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beneduce, E.; Matte, A.; De Falco, L.; Mbiandjeu, S.; Chiabrando, D.; Tolosano, E.; Federti, E.; Petrillo, S.; Mohandas, N.; Siciliano, A.; et al. Fyn kinase is a novel modulator of erythropoietin signaling and stress erythropoiesis. Am. J. Hematol. 2019, 94, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Svahn, J.; Lanza, T.; Rathbun, K.; Bagby, G.; Ravera, S.; Corsolini, F.; Pistorio, A.; Longoni, D.; Farruggia, P.; Dufour, C.; et al. p38 Mitogen-activated protein kinase inhibition enhances in vitro erythropoiesis of Fanconi anemia, complementation group A-deficient bone marrow cells. Exp. Hematol. 2015, 43, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Machado, P.; Manco, L.; Gomes, C.; Mendes, C.; Fernandes, N.; Salomé, G.; Sitoe, L.; Chibute, S.; Langa, J.; Ribeiro, L.; et al. Pyruvate kinase deficiency in sub-Saharan Africa: Identification of a highly frequent missense mutation (G829A;Glu277Lys) and association with malaria. PLoS ONE 2012, 7, e47071. [Google Scholar] [CrossRef]
- Remacha, Á.F.; Monter Rovira, A.; Esquirol Santfeliu, A.; Payán-Pernía, S.; Martino Bofarull, R.; García-Cadenas, I.; Brunet Mauri, S.; Sierra Gil, J. Microcytic anemia associated with mTOR or calcineurin inhibition: An unusual situation after allogeneic hematopoietic stem cell transplantation. Int. J. Lab. Hematol. 2020, 42, e141–e143. [Google Scholar] [CrossRef]
- Roux, P.P.; Topisirovic, I. Signaling Pathways Involved in the Regulation of mRNA Translation. Mol. Cell. Biol. 2018, 38, e00070-18. [Google Scholar] [CrossRef] [PubMed]
- Kamimae-Lanning, A.N.; Kurre, P. L-Leucine alleviates Diamond-Blackfan anemia. Blood 2012, 120, 2157–2158. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Schmidt, S.F.; Birsoy, K.; Tan, K.; Friedman, J.M. A critical role for mTORC1 in erythropoiesis and anemia. Elife 2014, 3, e01913. [Google Scholar] [CrossRef] [PubMed]
- Diekmann, F.; Rovira, J.; Diaz-Ricart, M.; Arellano, E.M.; Vodenik, B.; Jou, J.M.; Vives-Corrons, J.L.; Escolar, G.; Campistol, J.M. mTOR inhibition and erythropoiesis: Microcytosis or anaemia? Nephrol. Dial. Transplant. 2012, 27, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Zhang, S. Heme-regulated eIF2α kinase in erythropoiesis and hemoglobinopathies. Blood 2019, 134, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Geron, I.; Abrahamsson, A.E.; Barroga, C.F.; Kavalerchik, E.; Gotlib, J.; Hood, J.D.; Durocher, J.; Mak, C.C.; Noronha, G.; Soll, R.M.; et al. Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell 2008, 13, 321–330. [Google Scholar] [CrossRef]
- Ishitani, T.; Ishitani, S. Nemo-like kinase, a multifaceted cell signaling regulator. Cell. Signal. 2013, 25, 190–197. [Google Scholar] [CrossRef]
- Hu, P.; Nebreda, A.R.; Hanenberg, H.; Kinnebrew, G.H.; Ivan, M.; Yoder, M.C.; Filippi, M.D.; Broxmeyer, H.E.; Kapur, R. P38α/JNK signaling restrains erythropoiesis by suppressing Ezh2-mediated epigenetic silencing of Bim. Nat. Commun. 2018, 9, 3518. [Google Scholar] [CrossRef]
- Rubiolo, C.; Piazzolla, D.; Meissl, K.; Beug, H.; Huber, J.C.; Kolbus, A.; Baccarini, M. A balance between Raf-1 and Fas expression sets the pace of erythroid differentiation. Blood 2006, 108, 152–159. [Google Scholar] [CrossRef][Green Version]
- Saadatzadeh, M.R.; Bijangi-Vishehsaraei, K.; Kapur, R.; Haneline, L.S. Distinct roles of stress-activated protein kinases in Fanconi anemia-type C-deficient hematopoiesis. Blood 2009, 113, 2655–2660. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cichocki, F.; Felices, M.; McCullar, V.; Presnell, S.R.; Al-Attar, A.; Lutz, C.T.; Miller, J.S. Cutting edge: MicroRNA-181 promotes human NK cell development by regulating Notch signaling. J. Immunol. 2011, 187, 6171–6175. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.; Gao, L.; McClellan, S.; Finan, M.A.; Butler, T.W.; Owen, L.B.; Piazza, G.A.; Xi, Y. MiR-181 mediates cell differentiation by interrupting the Lin28 and let-7 feedback circuit. Cell Death Differ. 2012, 19, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Daams, R.; Massoumi, R. Nemo-Like Kinase in Development and Diseases: Insights from Mouse Studies. Int. J. Mol. Sci. 2020, 21, 9203. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkes, M.C.; Shibuya, A.; Sakamoto, K.M. Signaling Pathways That Regulate Normal and Aberrant Red Blood Cell Development. Genes 2021, 12, 1646. https://doi.org/10.3390/genes12101646
Wilkes MC, Shibuya A, Sakamoto KM. Signaling Pathways That Regulate Normal and Aberrant Red Blood Cell Development. Genes. 2021; 12(10):1646. https://doi.org/10.3390/genes12101646
Chicago/Turabian StyleWilkes, Mark C., Aya Shibuya, and Kathleen M. Sakamoto. 2021. "Signaling Pathways That Regulate Normal and Aberrant Red Blood Cell Development" Genes 12, no. 10: 1646. https://doi.org/10.3390/genes12101646
APA StyleWilkes, M. C., Shibuya, A., & Sakamoto, K. M. (2021). Signaling Pathways That Regulate Normal and Aberrant Red Blood Cell Development. Genes, 12(10), 1646. https://doi.org/10.3390/genes12101646