
Environmental Alterations during Embryonic Development: Studying the Impact of Stressors on Pluripotent Stem Cell-Derived Cardiomyocytes

 ,
,  ,
,
Abstract
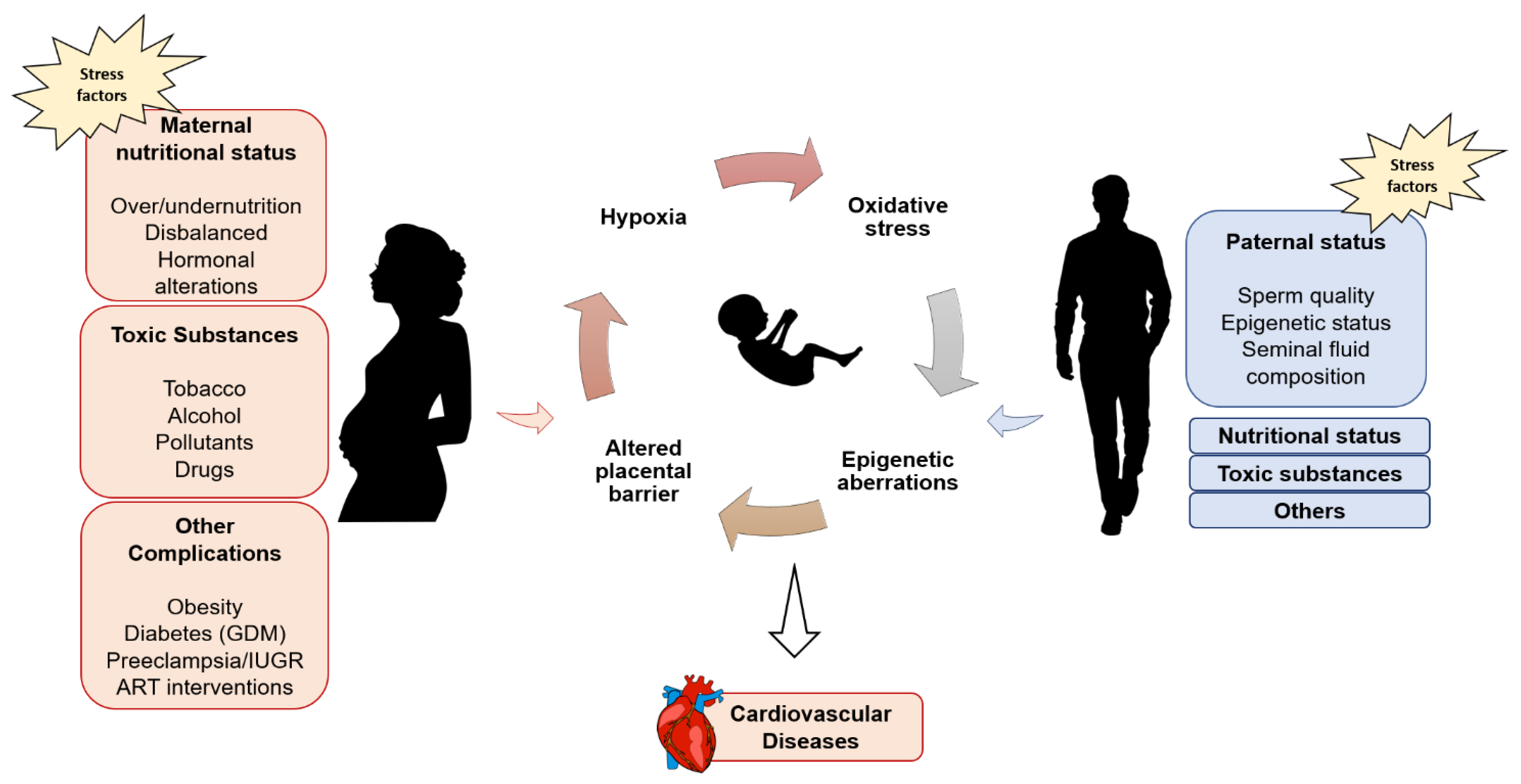
1. Introduction
2. The DOHaD Concept and Non-Communicable Diseases (NCDs)
3. Study of DOHAD: Observational Evidence
3.1. Human Studies
3.2. Animal Models
3.3. Novel Approaches to Model the DOHaD Concept In Vitro: Focus on Cardiovascular Diseases
4. Epigenetic Background of Cardiovascular Diseases
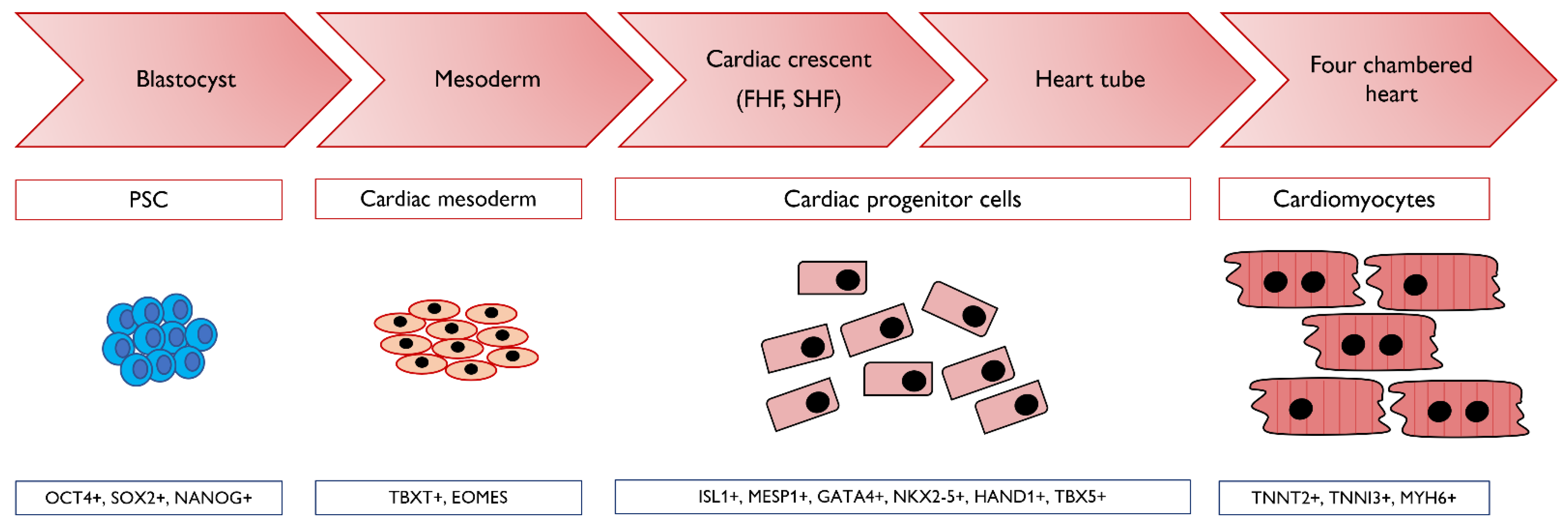
5. Modelling Cardio Myogenesis with Pluripotent Stem Cells
6. Studying the Effects of Environmental Factors on Cardiomyocytes Differentiation
6.1. Nutritional Effects on Cardiomyocytes Function
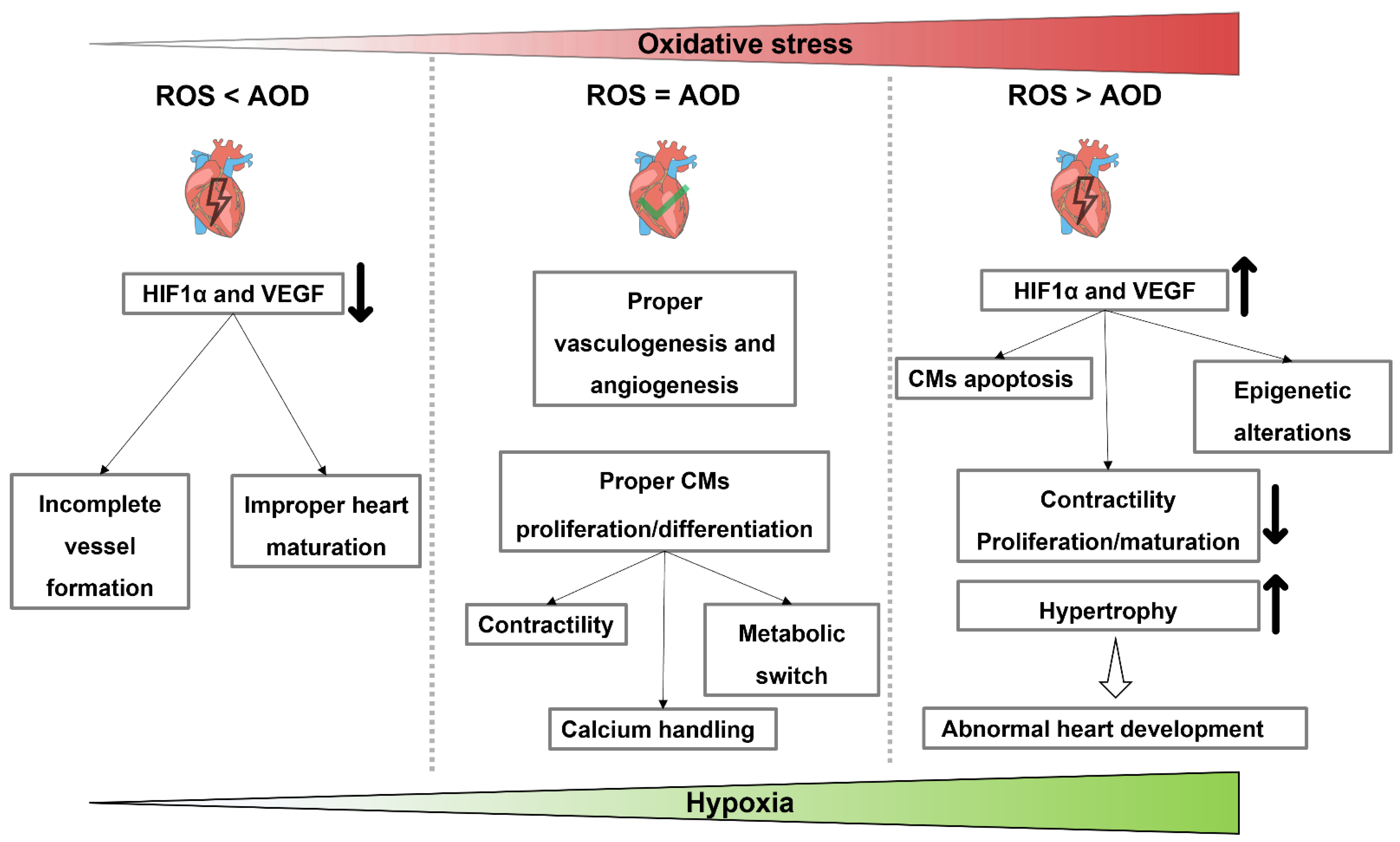
6.2. The Role of Hypoxia and Reactive Oxygen Species on Cardiomyocytes
6.3. Alcohol Consumption and Cigarette Smoking
6.4. Glucocorticoids
6.5. Chemical Exposure
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARE | antioxidant response element |
| ART | assisted reproductive technologies |
| ATP | adenosine triphosphate |
| BMI | Body Mass Index |
| CHD | congenital heart defects |
| CMs | cardiomyocytes |
| CVD | cardiovascular disease |
| DOHaD | Developmental Origins of Health and Disease |
| EBs | embryoid bodies |
| ECC | embryonal carcinoma cells |
| EZH2 | enhancer of zeste homolog 2 |
| eNOS | endothelial nitric oxide synthetase |
| ESC | embryonic stem cell |
| FFA | free fatty acids |
| GDM | gestational diabetes mellitus |
| HATs | histone acetyltransferases |
| HDACs | histone deacetylases |
| hESC-CMs | human embryonic stem cell-derived cardiomyocytes |
| hESCs | human embryonic stem cells |
| HFD | high-fat diet |
| HIFs | hypoxia-inducible factors |
| HIF1α | hypoxia-inducible factor 1α |
| hiPSC-CMs | human induced pluripotent stem cell-derived cardiomyocytes |
| hiPSCs | human induced pluripotent stem cells |
| HLHS | hypoplastic left heart syndrome |
| HMOX1 | heme oxygenase-1 |
| hPSC | human pluripotent stem cell |
| hPSC-CMs | human pluripotent stem cell-derived cardiomyocytes |
| ICSI | intracytoplasmic sperm injection |
| iPSC | induced pluripotent stem cell |
| IUGR | intra uterine growth restriction |
| IVF | in vitro fertilisation |
| JMJD2A | Jumonji Domain Containing 2A |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| LCFA | long-chain fatty acid |
| mESC | mouse embryonic stem cell |
| mESC-CMs | mouse embryonic stem cell-derived cardiomyocytes |
| miPSC-CMs | mouse induced pluripotent stem cell-derived cardiomyocytes |
| NCDs | Non-communicable diseases |
| NFE2L2/NRF2 | Nuclear factor erythroid 2-like 2 |
| NO | nitric oxide |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| PC | periconceptional |
| PCR | phosphocreatine |
| PRC2 | polycomb repressive complex 2 |
| PPARs | peroxisome proliferator-activated receptors |
| PSC-CMs | pluripotent stem cell-derived cardiomyocytes |
| PSCs | pluripotent stem cells |
| ROS | reactive oxygen species |
| SBP | systolic blood pressure |
| SCs | stem cell |
| SOD2 | superoxide dismutase-2 |
| T1DM | type 1 diabetes mellitus |
| T2DM | type 2 diabetes mellitus |
| TGF-β | transforming growth factor-β |
| WHO | World Health Organization |
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5-methylcytosine |
References
- Fleming, T.P.; Watkins, A.J.; Velazquez, M.A.; Mathers, J.C.; Prentice, A.M.; Stephenson, J.; Barker, M.; Saffery, R.; Yajnik, C.S.; Eckert, J.J.; et al. Origins of Lifetime Health Around the Time of Conception: Causes and Consequences. Obstet. Gynecol. Surv. 2018, 73, 555–557. [Google Scholar] [CrossRef]
- Velazquez, M.A.; Fleming, T.P.; Watkins, A.J. Periconceptional environment and the developmental origins of disease. J. Endocrinol. 2019, 242, T33–T49. [Google Scholar] [CrossRef] [PubMed]
- Yajnik, C.; Ganpule-Rao, A.; Limaye, T.; Rajgara, F. Developmental origins of non-communicable diseases. Proc. Indian Natl. Sci. Acad. 2016, 82, 1465–1475. [Google Scholar] [CrossRef]
- Mandy, M.; Nyirenda, M. Developmental Origins of Health and Disease: The relevance to developing nations. Int. Health 2018, 10, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.; Jacob, C.; Barker, M.; Fall, C.; Hanson, M.; Harvey, N.; Inskip, H.; Kumaran, K.; Cooper, C. Developmental Origins of Health and Disease: A Lifecourse Approach to the Prevention of Non-Communicable Diseases. Healthcare 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Gabory, A.; Attig, L.; Junien, C. Developmental programming and epigenetics 1–4. Am. J. Clin. Nutr. 2011, 94, 1943S–1952S. [Google Scholar] [CrossRef]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef]
- Zhu, Z.; Cao, F.; Li, X. Epigenetic Programming and Fetal Metabolic Programming. Front. Endocrinol. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Fleming, T.P.; Watkins, A.J.; Sun, C.; Velazquez, M.A.; Smyth, N.R.; Eckert, J.J. Do little embryos make big decisions? How maternal dietary protein restriction can permanently change an embryo’s potential, affecting adult health. Reprod. Fertil. Dev. 2015, 27, 684. [Google Scholar] [CrossRef]
- Eckert, J.J.; Porter, R.; Watkins, A.J.; Burt, E.; Brooks, S.; Leese, H.J.; Humpherson, P.G.; Cameron, I.T.; Fleming, T.P. Metabolic Induction and Early Responses of Mouse Blastocyst Developmental Programming following Maternal Low Protein Diet Affecting Life-Long Health. PLoS ONE 2012, 7, e52791. [Google Scholar] [CrossRef]
- Fleming, T.P.; Velazquez, M.A.; Eckert, J.J.; Lucas, E.S.; Watkins, A.J. Nutrition of females during the peri-conceptional period and effects on foetal programming and health of offspring. Anim. Reprod. Sci. 2012, 130, 193–197. [Google Scholar] [CrossRef]
- Gould, J.M.; Smith, P.J.; Airey, C.J.; Mort, E.J.; Airey, L.E.; Warricker, F.D.M.; Pearson-Farr, J.E.; Weston, E.C.; Gould, P.J.W.; Semmence, O.G.; et al. Mouse maternal protein restriction during preimplantation alone permanently alters brain neuron proportion and adult short-term memory. Proc. Natl. Acad. Sci. USA 2018, 115, E7398–E7407. [Google Scholar] [CrossRef]
- Kelleher, A.M.; Burns, G.W.; Behura, S.; Wu, G.; Spencer, T.E. Uterine glands impact uterine receptivity, luminal fluid homeostasis and blastocyst implantation. Sci. Rep. 2016, 6, 1–18. [Google Scholar] [CrossRef]
- Ye, X. Uterine Luminal Epithelium as the Transient Gateway for Embryo Implantation. Trends Endocrinol. Metab. 2020, 31, 165–180. [Google Scholar] [CrossRef]
- Swanson, L.D.; Bewtra, C. Increase in normal placental weights related to increase in maternal body mass index. J. Matern. Neonatal Med. 2008, 21, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Frias, A.E.; Morgan, T.K.; Evans, A.E.; Rasanen, J.; Oh, K.Y.; Thornburg, K.L.; Grove, K.L. Maternal High-Fat Diet Disturbs Uteroplacental Hemodynamics and Increases the Frequency of Stillbirth in a Nonhuman Primate Model of Excess Nutrition. Endocrinology 2011, 152, 2456–2464. [Google Scholar] [CrossRef]
- McMullen, S.; Mostyn, A. Animal models for the study of the developmental origins of health and disease. Proc. Nutr. Soc. 2009, 68, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.; Seki, Y.; Vuguin, P.M.; Charron, M.J. Animal models of in utero exposure to a high fat diet: A review. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Lurbe, E.; Ingelfinger, J. Developmental and Early Life Origins of Cardiometabolic Risk Factors: Novel Findings and Implications. Hypertension 2021, 77, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Huangfu, D. Human pluripotent stem cells: An emerging model in developmental biology. Development 2013, 140, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Osmond, C. Low birth weight and hypertension. BMJ 1988, 297, 134–135. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C.; Winter, P.D.; Margetts, B.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 334, 577–580. [Google Scholar] [CrossRef]
- Barker, D.J.P. The fetal and infant origins of disease. Eur. J. Clin. Investig. 1995, 25, 457–463. [Google Scholar] [CrossRef]
- World Health Organization. Chapter 1: Burden: Mortality, morbidity and risk factors. In Global Status Report on Non-Communicable Diseases; World Health Organization: Geneva, Switzerland, 2010; pp. 9–31. ISBN 9789240686458. [Google Scholar]
- World Health Organization. Noncommunicable Diseases Progress Monitor 2020; World Health Organization: Geneva, Switzerland, 2020; ISBN 9789240000490. [Google Scholar]
- Hanson, M.A.; Gluckman, P.D. Early developmental conditioning of later health and disease: Physiology or pathophysiology? Physiol. Rev. 2014, 94, 1027–1076. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.J.; Reynolds, R.M.; Hardy, D.B. Developmental origins of health and disease: Current knowledge and potential mechanisms. Nutr. Rev. 2017, 75, 951–970. [Google Scholar] [CrossRef]
- Faddy, M.J.; Gosden, M.D.; Gosden, R.G. A demographic projection of the contribution of assisted reproductive technologies to world population growth. Reprod. Biomed. Online 2018, 36, 455–458. [Google Scholar] [CrossRef]
- Sunde, A.; Brison, D.; Dumoulin, J.; Harper, J.; Lundin, K.; Magli, M.C.; Van Den Abbeel, E.; Veiga, A. Time to take human embryo culture seriously. Hum. Reprod. 2016, 31, 2174–2182. [Google Scholar] [CrossRef]
- Sinclair, K.D.; Singh, R. Modelling the developmental origins of health and disease in the early embryo. Theriogenology 2007, 67, 43–53. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C. Infant Mortality, Childhood Nutrition, and Ischaemic Heart Disease in England and Wales. Lancet 1986, 327, 1077–1081. [Google Scholar] [CrossRef]
- Ravelli, A.C.J.; Van Der Meulen, J.H.P.; Michels, R.P.J.; Osmond, C.; Barker, D.J.P.; Hales, C.N.; Bleker, O.P. Glucose tolerance in adults after prenatal exposure to famine. Lancet 1998, 351, 173–177. [Google Scholar] [CrossRef]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.J. Epidemiological evidence for the developmental origins of health and disease: Effects of prenatal undernutrition in humans. J. Endocrinol. 2019, 242, T135–T144. [Google Scholar] [CrossRef]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef]
- Wang, P.X.; Wang, J.J.; Lei, Y.X.; Xiao, L.; Luo, Z.C. Impact of Fetal and Infant Exposure to the Chinese Great Famine on the Risk of Hypertension in Adulthood. PLoS ONE 2012, 7, 1–8. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.V.; Eriksson, J.G.; Broekman, B.F.P. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef]
- Gonzalez, M.B.; Lane, M.; Knight, E.J.; Robker, R.L. Inflammatory markers in human follicular fluid correlate with lipid levels and Body Mass Index. J. Reprod. Immunol. 2018, 130, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Longmore, D.K.; Barr, E.L.M.; Lee, I.L.; Barzi, F.; Kirkwood, M.; Whitbread, C.; Hampton, V.; Graham, S.; Van Dokkum, P.; Connors, C.; et al. Maternal body mass index, excess gestational weight gain, and diabetes are positively associated with neonatal adiposity in the Pregnancy and Neonatal Diabetes Outcomes in Remote Australia (PANDORA) study. Pediatr. Obes. 2019, 14, 1–9. [Google Scholar] [CrossRef]
- Smith, J.; Cianflone, K.; Biron, S.; Hould, F.S.; Lebel, S.; Marceau, S.; Lescelleur, O.; Biertho, L.; Simard, S.; Kral, J.G.; et al. Effects of Maternal Surgical Weight Loss in Mothers on Intergenerational Transmission of Obesity. J. Clin. Endocrinol. Metab. 2009, 94, 4275–4283. [Google Scholar] [CrossRef]
- Snider, A.P.; Wood, J.R. Obesity induces ovarian inflammation and reduces oocyte quality. Reproduction 2019, 158, R79–R90. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E.J. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. Br. Med. J. 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, H.L.; Ozanne, S.E. Maternal diet-induced obesity and offspring cardiovascular health. J. Dev. Orig. Health Dis. 2013, 4, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Pettitt, D.J.; Hanson, R.L.; Imperatore, G.; Bennett, P.H.; Knowler, W.C. Birth weight, type 2 diabetes, and insulin resistance in Pima Indian children and young adults. Diabetes Care 1999, 22, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Bygren, L.O.; Kaati, G.; Edvinsson, S. Longevity determined by paternal ancestors’ nutrition during their slow growth period. Acta Biotheor. 2001, 49, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Lane, M.; Owens, J.A.; Bakos, H.W. Paternal obesity negatively affects male fertility and assisted reproduction outcomes: A systematic review and meta-analysis. Reprod. Biomed. Online 2015, 31, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Crean, A.J.; Bonduriansky, R. What is a paternal effect? Trends Ecol. Evol. 2014, 29, 554–559. [Google Scholar] [CrossRef]
- Curley, J.P.; Mashoodh, R.; Champagne, F.A. Epigenetics and the origins of paternal effects. Horm. Behav. 2011, 59, 306–314. [Google Scholar] [CrossRef]
- Kaati, G.; Bygren, L.O.; Edvinsson, S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur. J. Hum. Genet. 2002, 10, 682–688. [Google Scholar] [CrossRef]
- Kort, H.I.; Massey, J.B.; Elsner, C.W.; Mitchell-Leef, D.; Shapiro, D.B.; Witt, M.A.; Roudebush, W.E. Impact of body mass index values on sperm quantity and quality. J. Androl. 2006, 27, 450–452. [Google Scholar] [CrossRef]
- Pembrey, M.E.; Bygren, L.O.; Kaati, G.; Edvinsson, S.; Northstone, K.; Sjöström, M.; Golding, J. Sex-specific, male-line transgenerational responses in humans. Eur. J. Hum. Genet. 2006, 14, 159–166. [Google Scholar] [CrossRef]
- Tunc, O.; Bakos, H.W.; Tremellen, K. Impact of body mass index on seminal oxidative stress. Andrologia 2011, 43, 121–128. [Google Scholar] [CrossRef]
- Valenzuela-Alcaraz, B.; Crispi, F.; Bijnens, B.; Cruz-Lemini, M.; Creus, M.; Sitges, M.; Bartrons, J.; Civico, S.; Balasch, J.; Gratacós, E. Assisted reproductive technologies are associated with cardiovascular remodeling in utero that persists postnatally. Circulation 2013, 128, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Lazaraviciute, G.; Kauser, M.; Bhattacharya, S.; Haggarty, P.; Bhattacharya, S. A systematic review and meta-analysis of DNA methylation levels and imprinting disorders in children conceived by IVF/ICSI compared with children conceived spontaneously. Hum. Reprod. Update 2014, 20, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, H.; Moss, T.J.; Gatford, K.L.; Moritz, K.M.; Akison, L.; Fullston, T.; Hryciw, D.H.; Maloney, C.A.; Morris, M.J.; Wooldridge, A.L.; et al. A review of fundamental principles for animal models of DOHaD research: An Australian perspective. J. Dev. Orig. Health Dis. 2016, 7, 449–472. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, U.; Yasuhara, J.; Garg, V. In Vivo and In Vitro Genetic Models of Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2019, 13, a036764. [Google Scholar] [CrossRef] [PubMed]
- Blin, G.; Liand, M.; Mauduit, C.; Chehade, H.; Benahmed, M.; Simeoni, U.; Siddeek, B. Maternal exposure to high-fat diet induces long-term derepressive chromatin marks in the heart. Nutrients 2020, 12, 181. [Google Scholar] [CrossRef] [PubMed]
- Kwong, W.Y.; Wild, A.E.; Roberts, P.; Willis, A.C.; Fleming, T.P. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development 2000, 127, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.S.; Cooke, C.L.; Davidge, S.T. In utero origins of hypertension: Mechanisms and targets for therapy. Physiol. Rev. 2016, 96, 549–603. [Google Scholar] [CrossRef]
- Simeoni, U.; Armengaud, J.-B.; Siddeek, B.; Tolsa, J.-F. Perinatal Origins of Adult Disease. Neonatology 2018, 113, 393–399. [Google Scholar] [CrossRef]
- Sun, C.; Burgner, D.P.; Ponsonby, A.L.; Saffery, R.; Huang, R.C.; Vuillermin, P.J.; Cheung, M.; Craig, J.M. Effects of early-life environment and epigenetics on cardiovascular disease risk in children: Highlighting the role of twin studies. Pediatr. Res. 2013, 73, 523–530. [Google Scholar] [CrossRef]
- Vuguin, P.M. Animal Models for Small for Gestational Age and Fetal Programing of Adult Disease. Horm. Res. Paediatr. 2007, 68, 113–123. [Google Scholar] [CrossRef]
- Watkins, A.J.; Rubini, E.; Hosier, E.D.; Morgan, H.L. Paternal programming of offspring health. Early Hum. Dev. 2020, 150, 105185. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.J.; Wilkins, A.; Cunningham, C.; Perry, V.H.; Seet, M.J.; Osmond, C.; Eckert, J.J.; Torrens, C.; Cagampang, F.R.A.; Cleal, J.; et al. Low protein diet fed exclusively during mouse oocyte maturation leads to behavioural and cardiovascular abnormalities in offspring. J. Physiol. 2008, 586, 2231–2244. [Google Scholar] [CrossRef] [PubMed]
- Savoji, H.; Mohammadi, M.H.; Rafatian, N.; Toroghi, M.K.; Wang, E.Y.; Zhao, Y.; Korolj, A.; Ahadian, S.; Radisic, M. Cardiovascular disease models: A game changing paradigm in drug discovery and screening. Biomaterials 2019, 198, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Burrage, D.M.; Braddick, L.; Cleal, J.K.; Costello, P.; Noakes, D.E.; Hanson, M.A.; Green, L.R. The late gestation fetal cardiovascular response to hypoglycaemia is modified by prior peri-implantation undernutrition in sheep. J. Physiol. 2009, 587, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Fainberg, H.P.; Almond, K.L.; Li, D.; Rauch, C.; Bikker, P.; Symonds, M.E.; Mostyn, A. Impact of maternal dietary fat supplementation during gestation upon skeletal muscle in neonatal pigs. BMC Physiol. 2014, 14, 1–12. [Google Scholar] [CrossRef]
- Fan, L.; Lindsley, S.R.; Comstock, S.M.; Takahashi, D.L.; Evans, A.E.; He, G.W.; Thornburg, K.L.; Grove, K.L. Maternal high-fat diet impacts endothelial function in nonhuman primate offspring. Int. J. Obes. 2013, 37, 254–262. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Investig. 2009, 119, 323–335. [Google Scholar] [CrossRef]
- Torrens, C.; Snelling, T.H.; Chau, R.; Shanmuganathan, M.; Cleal, J.K.; Poore, K.R.; Noakes, D.E.; Poston, L.; Hanson, M.A.; Green, L.R. Effects of pre- and periconceptional undernutrition on arterial function in adult female sheep are vascular bed dependent. Exp. Physiol. 2009, 94, 1024–1033. [Google Scholar] [CrossRef]
- Xu, M.; Che, L.; Yang, Z.; Zhang, P.; Shi, J.; Li, J.; Lin, Y.; Fang, Z.; Che, L.; Feng, B.; et al. Effect of high fat dietary intake during maternal gestation on offspring ovarian health in a pig model. Nutrients 2016, 8, 498. [Google Scholar] [CrossRef]
- Rajabzadeh, N.; Fathi, E.; Farahzadi, R. Stem cell-based regenerative medicine. Stem Cell Investig. 2019, 6. [Google Scholar] [CrossRef]
- Schuldiner, M.; Yanuka, O.; Itskovitz-Eldor, J.; Melton, D.A.; Benvenisty, N. Effects of eight growth factors on the differentiation of cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 11307–11312. [Google Scholar] [CrossRef]
- Sharma, A.; Sances, S.; Workman, M.J.; Svendsen, C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 2020, 26, 309–329. [Google Scholar] [CrossRef]
- Stover, A.E.; Schwartz, P.H. The generation of embryoid bodies from feeder-based or feeder-free human pluripotent stem cell cultures. Methods Mol. Biol. 2011, 767, 391–398. [Google Scholar] [CrossRef]
- Thomson, J.A. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.J. Developmental plasticity and its relevance to assisted human reproduction. Hum. Reprod. 2018, 33, 546–552. [Google Scholar] [CrossRef]
- Kivimäki, M.; Steptoe, A. Effects of stress on the development and progression of cardiovascular disease. Nat. Rev. Cardiol. 2018, 15, 215–229. [Google Scholar] [CrossRef]
- Bakker, H.; Jaddoe, V.W.V. Cardiovascular and metabolic influences of fetal smoke exposure. Eur. J. Epidemiol. 2011, 26, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Leybovitz-Haleluya, N.; Wainstock, T.; Landau, D.; Sheiner, E. Maternal smoking during pregnancy and the risk of pediatric cardiovascular diseases of the offspring: A population-based cohort study with up to 18-years of follow up. Reprod. Toxicol. 2018, 78, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kettner, L.O.; Matthiesen, N.B.; Ramlau-Hansen, C.H.; Kesmodel, U.S.; Bay, B.; Henriksen, T.B. Fertility treatment and childhood type 1 diabetes mellitus: A nationwide cohort study of 565,116 live births. Fertil. Steril. 2016, 106, 1751–1756. [Google Scholar] [CrossRef]
- Huntriss, J.; Balen, A.H.; Sinclair, K.D.; Brison, D.R.; Picton, H.M. Epigenetics and Reproductive Medicine: Scientific Impact Paper No. 57. BJOG An Int. J. Obstet. Gynaecol. 2018, 125, e43–e54. [Google Scholar] [CrossRef]
- Symonds, M.E.; Stephenson, T.; Gardner, D.S.; Budge, H. Long-term effects of nutritional programming of the embryo and fetus: Mechanisms and critical windows. Reprod. Fertil. Dev. 2007, 19, 53–63. [Google Scholar] [CrossRef]
- Watkins, A.J.; Sinclair, K.D. Paternal low protein diet affects adult offspring cardiovascular and metabolic function in mice. Am. J. Physiol.-Heart Circ. Physiol. 2014, 306, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Eberle, C.; Kirchner, M.F.; Herden, R.; Stichling, S. Paternal metabolic and cardiovascular programming of their offspring: A systematic scoping review. PLoS ONE 2020, 15, e0244826. [Google Scholar] [CrossRef] [PubMed]
- Jarrell; Lennon; Jacot Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease. Diseases 2019, 7, 52. [CrossRef] [PubMed]
- Wu, J.; Izpisua Belmonte, J.C. Stem Cells: A Renaissance in Human Biology Research. Cell 2016, 165, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.R.; Gay, M.S.; Zhang, L. Epigenetic mechanisms in heart development and disease. Drug Discov. Today 2015, 20, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Soler-Botija, C.; Gálvez-Montón, C.; Bayés-Genís, A. Epigenetic Biomarkers in Cardiovascular Diseases. Front. Genet. 2019, 10, 1–31. [Google Scholar] [CrossRef]
- Ordovás, J.M.; Smith, C.E. Epigenetics and cardiovascular disease. Nat. Rev. Cardiol. 2010, 7, 510–519. [Google Scholar] [CrossRef]
- Moore-Morris, T.; van Vliet, P.P.; Andelfinger, G.; Puceat, M. Role of epigenetics in cardiac development and congenital diseases. Physiol. Rev. 2018, 98, 2453–2475. [Google Scholar] [CrossRef]
- Ambra, R.; Manca, S.; Palumbo, M.C.; Leoni, G.; Natarelli, L.; De Marco, A.; Consoli, A.; Pandolfi, A.; Virgili, F. Transcriptome analysis of human primary endothelial cells (HUVEC) from umbilical cords of gestational diabetic mothers reveals candidate sites for an epigenetic modulation of specific gene expression. Genomics 2014, 103, 337–348. [Google Scholar] [CrossRef]
- Guo, R.; Nair, S. BBA—Molecular Basis of Disease Role of microRNA in diabetic cardiomyopathy: From mechanism to. BBA-Mol. Basis Dis. 2017, 1863, 2070–2077. [Google Scholar] [CrossRef]
- Lock, M.C.; Botting, K.J.; Tellam, R.L.; Brooks, D.; Morrison, J.L. Adverse intrauterine environment and cardiac miRNA expression. Int. J. Mol. Sci. 2017, 18, 2628. [Google Scholar] [CrossRef]
- Ruchat, S.M.; Houde, A.A.; Voisin, G.; St-Pierre, J.; Perron, P.; Baillargeon, J.P.; Gaudet, D.; Hivert, M.F.; Brisson, D.; Bouchard, L. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics 2013, 8, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Siddeek, B.; Mauduit, C.; Chehade, H.; Blin, G.; Liand, M.; Chindamo, M.; Benahmed, M.; Simeoni, U. Long-term impact of maternal high-fat diet on offspring cardiac health: Role of micro-RNA biogenesis. Cell Death Discov. 2019, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Grüning, B.A.; Kranzhöfer, D.; Schneider, P.; Nührenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef]
- Rommel, C.; Hein, L. Four Dimensions of the Cardiac Myocyte Epigenome: From Fetal to Adult Heart. Curr. Cardiol. Rep. 2020, 22, 26. [Google Scholar] [CrossRef]
- Ameer, S.S.; Hossain, M.B.; Knöll, R. Epigenetics and heart failure. Int. J. Mol. Sci. 2020, 21, 9019. [Google Scholar] [CrossRef]
- Gilsbach, R.; Preissl, S.; Grüning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Würch, A.; Bönisch, U.; Günther, S.; Backofen, R.; et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef]
- Long, F.; Wang, Q.; Yang, D.; Zhu, M.; Wang, J.; Zhu, Y.Z.; Liu, X. Targeting JMJD3 histone demethylase mediates cardiac fibrosis and cardiac function following myocardial infarction. Biochem. Biophys. Res. Commun. 2020, 528, 671–677. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Tran, T.A.T.; Wang, M.; Ranek, M.J.; Kokkonen-Simon, K.M.; Gao, J.; Luo, X.; Tan, W.; Kyrychenko, V.; Liao, L.; et al. Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Guénantin, A.C.; Jebeniani, I.; Leschik, J.; Watrin, E.; Bonne, G.; Vignier, N.; Pucéat, M. Targeting the histone demethylase LSD1 prevents cardiomyopathy in a mouse model of laminopathy. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.J.; Chen, H.Z.; Wang, L.; Liu, D.P.; Hill, J.A.; Liu, Z.P. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J. Clin. Investig. 2011, 121, 2447–2456. [Google Scholar] [CrossRef] [PubMed]
- Rosales, W.; Lizcano, F. The Histone Demethylase JMJD2A Modulates the Induction of Hypertrophy Markers in iPSC-Derived Cardiomyocytes. Front. Genet. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Chapple, S.J.; Puszyk, W.M.; Mann, G.E. Keap1-NRF2 regulated redox signaling in utero: Priming of disease susceptibility in offspring. Free Radic. Biol. Med. 2015, 88, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.B.; Jones, T.A.; Herron, T.J.; Patel, S.R.; Day, S.M.; Noujaim, S.F.; Milstein, M.L.; Klos, M.; Furspan, P.B.; Jalife, J.; et al. Loss of H3K4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J. Clin. Investig. 2011, 121, 2641–2650. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.J.; Chen, M.; Xue, Q.; Xiao, D.; Zhang, L. Chronic prenatal hypoxia induces epigenetic programming of PKCε gene repression in rat hearts. Circ. Res. 2010, 107, 365–373. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the fetal gene program: A suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic gene expression in fetal and failing human heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef]
- Delgado-Olguín, P.; Huang, Y.; Li, X.; Christodoulou, D.; Seidman, C.E.; Seidman, J.G.; Tarakhovsky, A.; Bruneau, B.G. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat. Genet. 2012, 44, 343–347. [Google Scholar] [CrossRef]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic modifications in cardiovascular aging and diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- He, A.; Ma, Q.; Cao, J.; Von Gise, A.; Zhou, P.; Xie, H.; Zhang, B.; Hsing, M.; Christodoulou, D.C.; Cahan, P.; et al. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ. Res. 2012, 110, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, K.T.; Jones, D.C.; Sperber, H.; Madan, A.; Fischer, K.A.; Rodriguez, M.L.; Pabon, L.; Zhu, W.Z.; Tulloch, N.L.; Yang, X.; et al. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, E2785–E2794. [Google Scholar] [CrossRef]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.J.; Matkovich, S.J.; Dorn, G.W.; Van Rooij, E.; Olson, E.N. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Abdellatif, M. Micrornas in development and disease. Physiol. Rev. 2011, 91, 827–887. [Google Scholar] [CrossRef] [PubMed]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef]
- Piquereau, J.; Ventura-Clapier, R. Maturation of cardiac energy metabolism during perinatal development. Front. Physiol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Nakano, H.; Minami, I.; Braas, D.; Pappoe, H.; Wu, X.; Sagadevan, A.; Vergnes, L.; Fu, K.; Morselli, M.; Dunham, C.; et al. Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. Elife 2017, 6, e29330. [Google Scholar] [CrossRef] [PubMed]
- Pohjoismäki, J.L.; Goffart, S. The role of mitochondria in cardiac development and protection. Free Radic. Biol. Med. 2017, 106, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defned generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Fonoudi, H.; Bosman, A. Turning potential into action: Using pluripotent stem cells to understand heart development and function in health and disease. Stem Cells Transl. Med. 2017, 6, 1452–1457. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leitolis, A.; Robert, A.W.; Pereira, I.T.; Correa, A.; Stimamiglio, M.A. Cardiomyogenesis Modeling Using Pluripotent Stem Cells: The Role of Microenvironmental Signaling. Front. Cell Dev. Biol. 2019, 7, 1–20. [Google Scholar] [CrossRef]
- Atmanli, A.; Domian, I.J. Recreating the Cardiac Microenvironment in Pluripotent Stem Cell Models of Human Physiology and Disease. Trends Cell Biol. 2017, 27, 352–364. [Google Scholar] [CrossRef]
- Hamad, S.; Derichsweiler, D.; Papadopoulos, S.; Nguemo, F.; Šarić, T.; Sachinidis, A.; Brockmeier, K.; Hescheler, J.; Boukens, B.J.; Pfannkuche, K. Generation of human induced pluripotent stem cell-derived cardiomyocytes in 2D monolayer and scalable 3D suspension bioreactor cultures with reduced batch-to-batch variations. Theranostics 2019, 9, 7222–7238. [Google Scholar] [CrossRef]
- Masumoto, H.; Ikuno, T.; Takeda, M.; Fukushima, H.; Marui, A.; Katayama, S.; Shimizu, T.; Ikeda, T.; Okano, T.; Sakata, R.; et al. Human iPS cell-engineered cardiac tissue sheets with cardiomyocytes and vascular cells for cardiac regeneration. Sci. Rep. 2014, 4, 6716. [Google Scholar] [CrossRef]
- Lu, T.-Y.; Lin, B.; Kim, J.; Sullivan, M.; Tobita, K.; Salama, G.; Yang, L. Repopulation of decellularized mouse heart with human induced pluripotent stem cell-derived cardiovascular progenitor cells. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Rajabi, S.; Pahlavan, S.; Ashtiani, M.K.; Ansari, H.; Abbasalizadeh, S.; Sayahpour, F.A.; Varzideh, F.; Kostin, S.; Aghdami, N.; Braun, T.; et al. Human embryonic stem cell-derived cardiovascular progenitor cells efficiently colonize in bFGF-tethered natural matrix to construct contracting humanized rat hearts. Biomaterials 2018, 154, 99–112. [Google Scholar] [CrossRef]
- Jha, R.; Xu, R.H.; Xu, C. Efficient differentiation of cardiomyocytes from human pluripotent stem cells with growth factors. In Cardiomyocytes: Methods and Protocols; Springer: New York, NY, USA, 2015; Volume 1299, pp. 115–131. ISBN 9781493925728. [Google Scholar]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- Scuderi, G.J.; Butcher, J. Naturally engineered maturation of cardiomyocytes. Front. Cell Dev. Biol. 2017, 5, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; Van Der Meer, P.; Serra, M.; et al. Metabolic maturation of human pluripotent stem cellderived cardiomyocytes by inhibition of HIF1α and LDHA. Circ. Res. 2018, 123, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Hirata, M.; Yamaoka, T. Effect of stem cell niche elasticity/ECM protein on the self-beating cardiomyocyte differentiation of induced pluripotent stem (iPS) cells at different stages. Acta Biomater. 2018, 65, 44–52. [Google Scholar] [CrossRef]
- Ruan, J.-L.; Tulloch, N.L.; Razumova, M.V.; Saiget, M.; Muskheli, V.; Pabon, L.; Reinecke, H.; Regnier, M.; Murry, C.E. Mechanical Stress Conditioning and Electrical Stimulation Promote Contractility and Force Maturation of Induced Pluripotent Stem Cell-Derived Human Cardiac Tissue. Circulation 2016, 134, 1557–1567. [Google Scholar] [CrossRef]
- Mogi, A.; Takei, S.; Shimizu, H.; Miura, H.; Tomotsune, D.; Sasaki, K. Effects of fluid dynamic forces created by rotary orbital suspension culture on cardiomyogenic differentiation of human embryonic stem cells. J. Med. Biol. Eng. 2014, 34, 101–108. [Google Scholar] [CrossRef]
- Choy, M.K.; Javierre, B.M.; Williams, S.G.; Baross, S.L.; Liu, Y.; Wingett, S.W.; Akbarov, A.; Wallace, C.; Freire-Pritchett, P.; Rugg-Gunn, P.J.; et al. Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS regions to cardiac gene networks. Nat. Commun. 2018, 9, 2526. [Google Scholar] [CrossRef]
- Montefiori, L.E.; Sobreira, D.R.; Sakabe, N.J.; Aneas, I.; Joslin, A.C.; Hansen, G.T.; Bozek, G.; Moskowitz, I.P.; McNally, E.M.; Nóbrega, M.A. A promoter interaction map for cardiovascular disease genetics. Elife 2018, 7, 1–35. [Google Scholar] [CrossRef]
- Bertero, A.; Fields, P.A.; Ramani, V.; Bonora, G.; Yardimci, G.G.; Reinecke, H.; Pabon, L.; Noble, W.S.; Shendure, J.; Murry, C.E. Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef]
- Bertero, A.; Rosa-Garrido, M. Three-dimensional chromatin organization in cardiac development and disease. J. Mol. Cell. Cardiol. 2021, 151, 89–105. [Google Scholar] [CrossRef]
- Biermann, M.; Cai, W.; Lang, D.; Hermsen, J.; Profio, L.; Zhou, Y.; Czirok, A.; Isai, D.G.; Napiwocki, B.N.; Rodriguez, A.M.; et al. Epigenetic Priming of Human Pluripotent Stem Cell-Derived Cardiac Progenitor Cells Accelerates Cardiomyocyte Maturation. Stem Cells 2019, 37, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.C.; Banovich, N.E.; Sarkar, A.; Stephens, M.; Gilad, Y. Dynamic effects of genetic variation on gene expression revealed following hypoxic stress in cardiomyocytes. Elife 2021, 10, e57345. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.; Brueckner, M.; Chung, W.; Goldmuntz, E.; Kaltman, J.; Kaski, J.P.; Kim, R.; Kline, J.; Mercer-Rosa, L.; Porter, G.; et al. The congenital heart disease genetic network study: Rationale, design, and early results. Circ. Res. 2013, 112, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Martewicz, S.; Magnussen, M.; Elvassore, N. Beyond Family: Modeling Non-hereditary Heart Diseases with Human Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Physiol. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- De Jong, K.A.; Barrand, S.; Wood-Bradley, R.J.; de Almeida, D.L.; Czeczor, J.K.; Lopaschuk, G.D.; Armitage, J.A.; McGee, S.L. Maternal high fat diet induces early cardiac hypertrophy and alters cardiac metabolism in Sprague Dawley rat offspring. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 600–609. [Google Scholar] [CrossRef]
- Xue, Q.; Chen, F.; Zhang, H.; Liu, Y.; Chen, P.; Patterson, A.J.; Luo, J. Maternal high-fat diet alters angiotensin II receptors and causes changes in fetal and neonatal rats†. Biol. Reprod. 2019, 100, 1193–1203. [Google Scholar] [CrossRef]
- Ma, X.M.; Shi, Q.Y.; Zhao, Y.X. Maternal exposure to a high-fat diet showed unfavorable effects on the body weight, apoptosis and morphology of cardiac myocytes in offspring. Arch. Gynecol. Obstet. 2020, 301, 837–844. [Google Scholar] [CrossRef]
- Watkins, A.J.; Lucas, E.S.; Wilkins, A.; Cagampang, F.R.A.; Fleming, T.P. Maternal Periconceptional and Gestational Low Protein Diet Affects Mouse Offspring Growth, Cardiovascular and Adipose Phenotype at 1 Year of Age. PLoS ONE 2011, 6, e28745. [Google Scholar] [CrossRef]
- Asopa, S.; Cagampang, F.R.; Anthony, F.W.; Lanham, S.A.; Schneider, J.E.; Ohri, S.K.; Hanson, M.A. Effect of a low-protein diet during pregnancy on expression of genes involved in cardiac hypertrophy in fetal and adult mouse offspring. J. Dev. Orig. Health Dis. 2010, 1, 371–375. [Google Scholar] [CrossRef]
- Gray, C.; Li, M.; Patel, R.; Reynolds, C.M.; Vickers, M.H. Let-7 miRNA Profiles Are Associated With the Reversal of Left Ventricular Hypertrophy and Hypertension in Adult Male Offspring From Mothers Undernourished During Pregnancy After Preweaning Growth Hormone Treatment. Endocrinology 2014, 155, 4808–4817. [Google Scholar] [CrossRef]
- Velazquez, M.A.; Sheth, B.; Smith, S.J.; Eckert, J.J.; Osmond, C.; Fleming, T.P. Insulin and branched-chain amino acid depletion during mouse preimplantation embryo culture programmes body weight gain and raised blood pressure during early postnatal life. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 590–600. [Google Scholar] [CrossRef]
- Corstius, H.B.; Zimanyi, M.A.; Maka, N.; Herath, T.; Thomas, W.; Van Der Laarse, A.; Wreford, N.G.; Black, M.J. Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr. Res. 2005, 57, 796–800. [Google Scholar] [CrossRef]
- Cheema, K.K.; Dent, M.R.; Saini, H.K.; Aroutiounova, N.; Tappia, P.S. Prenatal exposure to maternal undernutrition induces adult cardiac dysfunction. Br. J. Nutr. 2005, 93, 471–477. [Google Scholar] [CrossRef]
- Aroutiounova, N.; Fandrich, R.; Kardami, E.; Tappia, P.S. Prenatal exposure to maternal low protein diet suppresses replicative potential of myocardial cells. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, N.; Brazil, D.P.; McAuliffe, F. Fetal cardiac effects of maternal hyperglycemia during pregnancy. Birth Defects Res. Part A-Clin. Mol. Teratol. 2009, 85, 523–530. [Google Scholar] [CrossRef]
- Balistreri, M.; Davis, J.A.; Campbell, K.F.; Da Rocha, A.M.; Treadwell, M.C.; Herron, T.J. Effect of Glucose on 3D Cardiac Microtissues Derived from Human Induced Pluripotent Stem Cells. Pediatr. Cardiol. 2017, 38, 1575–1582. [Google Scholar] [CrossRef]
- Yu, Y.; Arah, O.A.; Liew, Z.; Cnattingius, S.; Olsen, J.; Sørensen, H.T.; Qin, G.; Li, J. Maternal diabetes during pregnancy and early onset of cardiovascular disease in offspring: Population based cohort study with 40 years of follow-up. BMJ 2019, 367, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Smoak, I.W. Hypoglycemia and Embryonic heart development. Front. Biosci. 2002, 7, 307–318. [Google Scholar] [CrossRef]
- Ng, K.M.; Lau, Y.M.; Dhandhania, V.; Cai, Z.J.; Lee, Y.K.; Lai, W.H.; Tse, H.F.; Siu, C.W. Empagliflozin Ammeliorates High Glucose Induced-Cardiac Dysfuntion in Human iPSC-Derived Cardiomyocytes. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Oligschlaeger, Y.; Geraets, I.; Liu, Y.; Zhu, X.; Li, J.; Nabben, M.; Coumans, W.; Luiken, J.J.F.P.; Glatz, J.F.C.; et al. 2-Arachidonoylglycerol ameliorates inflammatory stress-induced insulin resistance in cardiomyocytes. J. Biol. Chem. 2017, 292, 7105–7114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Steinbusch, L.K.M.; Nabben, M.; Kapsokalyvas, D.; Van Zandvoort, M.; Schönleitner, P.; Antoons, G.; Simons, P.J.; Coumans, W.A.; Geomini, A.; et al. Palmitate-induced vacuolar-type H+-ATPase inhibition feeds forward into insulin resistance and contractile dysfunction. Diabetes 2017, 66, 1521–1534. [Google Scholar] [CrossRef] [PubMed]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Nelson, A.; Lamba, D.A.; Reh, T.A.; Ware, C.; Ruohola-Baker, H. Hypoxia induces re-entry of committed cells into pluripotency. Stem Cells 2013, 31, 1737–1748. [Google Scholar] [CrossRef]
- Ng, K.M.; Lee, Y.K.; Chan, Y.C.; Lai, W.H.; Fung, M.L.; Li, R.A.; Siu, C.W.; Tse, H.F. Exogenous expression of HIF-1α promotes cardiac differentiation of embryonic stem cells. J. Mol. Cell. Cardiol. 2010, 48, 1129–1137. [Google Scholar] [CrossRef]
- Dunwoodie, S.L. The Role of Hypoxia in Development of the Mammalian Embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef]
- J. Patterson, A.; Zhang, L. Hypoxia and Fetal Heart Development. Curr. Mol. Med. 2010, 10, 653–666. [Google Scholar] [CrossRef]
- Bae, S.; Xiao, Y.; Li, G.; Casiano, C.A.; Zhang, L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am. J. Physiol.-Heart Circ. Physiol. 2003, 285, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Medley, T.L.; Furtado, M.; Lam, N.T.; Idrizi, R.; Williams, D.; Verma, P.J.; Costa, M.; Kaye, D.M. Effect of oxygen on cardiac differentiation in mouse iPS cells: Role of hypoxia inducible factor-1 and Wnt/β-catenin signaling. PLoS ONE 2013, 8, 1–8. [Google Scholar] [CrossRef]
- Gaber, N.; Gagliardi, M.; Patel, P.; Kinnear, C.; Zhang, C.; Chitayat, D.; Shannon, P.; Jaeggi, E.; Tabori, U.; Keller, G.; et al. Fetal reprogramming and senescence in hypoplastic left heart syndrome and in human pluripotent stem cells during cardiac differentiation. Am. J. Pathol. 2013, 183, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Yoshida, M.; Tarui, S.; Hirata, M.; Nagai, Y.; Kasahara, S.; Naruse, K.; Ito, H.; Sano, S.; Oh, H. Directed Differentiation of Patient-Specific Induced Pluripotent Stem Cells Identifies the Transcriptional Repression and Epigenetic Modification of NKX2-5, HAND1, and NOTCH1 in Hypoplastic Left Heart Syndrome. PLoS ONE 2014, 9, e102796. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, L.; Wang, C. Prenatal hypoxia-induced epigenomic and transcriptomic reprogramming in rat fetal and adult offspring hearts. Sci. Data 2019, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Bae, S.; Zhang, L. Effect of prenatal hypoxia on heat stress-mediated cardioprotection in adult rat heart. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H1712–H1719. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.B.; Hwang, K.A.; Choi, K.C. Prenatal toxicity of the environmental pollutants on neuronal and cardiac development derived from embryonic stem cells. Reprod. Toxicol. 2019, 90, 15–23. [Google Scholar] [CrossRef]
- Zheng, F.; Gonçalves, F.M.; Abiko, Y.; Li, H.; Kumagai, Y.; Aschner, M. Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Engineer, A.; Saiyin, T.; Greco, E.R.; Feng, Q. Say NO to ROS: Their roles in embryonic heart development and pathogenesis of congenital heart defects in maternal diabetes. Antioxidants 2019, 8, 436. [Google Scholar] [CrossRef]
- Chandel, N.S.; Mcclintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-inducible Factor-1α during Hypoxia. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef]
- Liang, J.; Wu, M.; Chen, C.; Mai, M.; Huang, J.; Zhu, P. Review Article Roles of Reactive Oxygen Species in Cardiac Differentiation, Reprogramming, and Regenerative Therapies. Oxidative Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Li, T.; Zhang, X.; Jiang, K.; Liu, J.; Liu, Z. Dural effects of oxidative stress on cardiomyogenesis via Gata4 transcription and protein ubiquitination article. Cell Death Dis. 2018, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.M.; Maltagliati, A.J. NRF2 at the heart of oxidative stress and cardiac protection. Physiol. Genomics 2018, 50, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wang, X.; Jiang, S.; Ru, N.; Jiao, B.; Wang, Y.; Yu, Z.; Wang, X. Elevated ROS depress mitochondrial oxygen utilization efficiency in cardiomyocytes during acute hypoxia. Pflugers Archiv-Eur. J. Physiol. 2020, 472, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Yan, M.S.; Matouk, C.C.; St. Bernard, R.; Ho, J.J.D.; Gavryushova, A.; Srivastava, D.; Marsfen, P.A. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J. Biol. Chem. 2010, 285, 810–826. [Google Scholar] [CrossRef]
- Crespo, F.L.; Sobrado, V.R.; Gomez, L.; Cervera, A.M.; McCreath, K.J. Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells 2010, 28, 1132–1142. [Google Scholar] [CrossRef]
- Murray, T.V.A.; Ahmad, A.; Brewer, A.C. Reactive oxygen at the heart of metabolism. Trends Cardiovasc. Med. 2014, 24, 113–120. [Google Scholar] [CrossRef]
- Momtahan, N.; Crosby, C.O.; Zoldan, J. The Role of Reactive Oxygen Species in In Vitro Cardiac Maturation. Trends Mol. Med. 2019, 25, 482–493. [Google Scholar] [CrossRef]
- De Giusti, V.C.; Correa, M.V.; Villa-Abrille, M.C.; Beltrano, C.; Yeves, A.M.; de Cingolani, G.E.C.; Cingolani, H.E.; Aiello, E.A. The positive inotropic effect of endothelin-1 is mediated by mitochondrial reactive oxygen species. Life Sci. 2008, 83, 264–271. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 60–67. [Google Scholar] [CrossRef]
- Földes, G.; Matsa, E.; Kriston-Vizi, J.; Leja, T.; Amisten, S.; Kolker, L.; Kodagoda, T.; Dolatshad, N.F.; Mioulane, M.; Vauchez, K.; et al. Aberrant α-adrenergic hypertrophic response in Cardiomyocytes from human induced pluripotent cells. Stem Cell Rep. 2014, 3, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Rampoldi, A.; Singh, M.; Wu, Q.; Duan, M.; Jha, R.; Maxwell, J.T.; Bradner, J.M.; Zhang, X.; Saraf, A.; Miller, G.W.; et al. Cardiac toxicity from ethanol exposure in human-induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. 2019, 169, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Umoh, N.A.; Walker, R.K.; Al-Rubaiee, M.; Jeffress, M.A.; Haddad, G.E. Acute Alcohol Modulates Cardiac Function as PI3K/Akt Regulates Oxidative Stress. Alcohol. Clin. Exp. Res. 2014, 38, 1847–1864. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Jin, C.M.; Li, S.S.; Zhang, F.M.; Yuan, L.; Li, W.M.; Sang, Y.; Li, S.; Zhou, L.J. Chronic alcohol intake-induced oxidative stress and apoptosis: Role of CYP2E1 and calpain-1 in alcoholic cardiomyopathy. Mol. Cell. Biochem. 2012, 359, 283–292. [Google Scholar] [CrossRef]
- Ramprasath, T.; Vasudevan, V.; Sasikumar, S.; Puhari, S.; Saso, L.; Selvam, G. Regression of Oxidative Stress by Targeting eNOS and NRF2/ARE Signaling: A Guided Drug Target for Cardiovascular Diseases. Curr. Top. Med. Chem. 2015, 15, 857–871. [Google Scholar] [CrossRef]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The Role of NRF2 in Cardiovascular Function and Disease. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, Q. NOing the heart: Role of nitric oxide synthase-3 in heart development. Differentiation 2012, 84, 54–61. [Google Scholar] [CrossRef]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS Uncoupling in Cardiovascular Diseases—The Role of Oxidative Stress and Inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef]
- Yang, H.; Kuhn, C.; Kolben, T.; Ma, Z.; Lin, P.; Mahner, S.; Jeschke, U.; von Schönfeldt, V. Early life oxidative stress and long-lasting cardiovascular effects on offspring conceived by assisted reproductive technologies: A review. Int. J. Mol. Sci. 2020, 21, 5175. [Google Scholar] [CrossRef]
- Kim, G.H.; Ryan, J.J.; Archer, S.L. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid. Redox Signal. 2013, 18, 1920–1936. [Google Scholar] [CrossRef]
- West, N.A.; Kechris, K.; Dabelea, D. Exposure to Maternal Diabetes in Utero and DNA Methylation Patterns in the Offspring. Immunometabolism 2013, 1, 1–9. [Google Scholar] [CrossRef]
- Liu, Z.Z.; Zhao, X.Z.; Zhang, X.S.; Zhang, M. Promoter DNA demethylation of Keap1 gene in diabetic cardiomyopathy. Int. J. Clin. Exp. Pathol. 2014, 7, 8756–8762. [Google Scholar]
- Rexhaj, E.; Sartori, C.; Scherrer, U. Shortened life span in mice generated by in vitro fertilization. FASEB J. 2013, 27, 5052–5060. [Google Scholar] [CrossRef]
- Basu, M.; Zhu, J.-Y.; LaHaye, S.; Majumdar, U.; Jiao, K.; Han, Z.; Garg, V. Epigenetic mechanisms underlying maternal diabetes-associated risk of congenital heart disease. JCI Insight 2017, 2, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Barandalla, M.; Shi, H.; Xiao, H.; Colleoni, S.; Galli, C.; Lio, P.; Trotter, M.; Lazzari, G. Global gene expression profiling and senescence biomarker analysis of hESC exposed to H2O2 induced non-cytotoxic oxidative stress. Stem Cell Res. Ther. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- M, B.; S, C. Differential Response of Human Embryonic Stem and Somatic Cells to Non-Cytotoxic Hydrogen Peroxide Exposure: An Attempt to Model In Vitro the Effects of Oxidative Stress on the Early Embryo. Cell Dev. Biol. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Goh, J.M.; Bensley, J.G.; Kenna, K.; Sozo, F.; Bocking, A.D.; Brien, J.; Walker, D.; Harding, R.; Black, M.J. Alcohol exposure during late gestation adversely affects myocardial development with implications for postnatal cardiac function. Am. J. Physiol.-Heart Circ. Physiol. 2011, 300, 645–651. [Google Scholar] [CrossRef]
- Guo, H.; Tian, L.; Zhang, J.Z.; Kitani, T.; Paik, D.T.; Lee, W.H.; Wu, J.C. Single-Cell RNA Sequencing of Human Embryonic Stem Cell Differentiation Delineates Adverse Effects of Nicotine on Embryonic Development. Stem Cell Rep. 2019, 12, 772–786. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, P.; Ramiro-Cortijo, D.; Reyes-Hernández, C.G.; López de Pablo, A.L.; Carmen González, M.; Arribas, S.M. Implication of oxidative stress in fetal programming of cardiovascular disease. Front. Physiol. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Santos, C.X.C.; Anilkumar, N.; Zhang, M.; Brewer, A.C.; Shah, A.M. Redox signaling in cardiac myocytes. Free Radic. Biol. Med. 2011, 50, 777–793. [Google Scholar] [CrossRef]
- Wang, Q.; Song, J.; Liu, Y.; Zhao, X. Involvement of Wnt pathway in ethanol-induced inhibition of mouse embryonic stem cell differentiation. Alcohol 2017, 58, 13–18. [Google Scholar] [CrossRef]
- Peng, B.; Han, X.; Peng, C.; Luo, X.; Deng, L.; Huang, L. G9α-dependent histone H3K9me3 hypomethylation promotes overexpression of cardiomyogenesis-related genes in foetal mice. J. Cell. Mol. Med. 2020, 24, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.M.; Dervan, L.A.; Reiger, S.; Buddhe, S.; Schwartz, S.M. Risk of congenital heart defects in the offspring of smoking mothers: A population-based study. J. Pediatr. 2015, 166, 978–984.e2. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Zhou, R.; Feng, Y.; Wang, Y. Mainstream smoke and sidestream smoke affect the cardiac differentiation of mouse embryonic stem cells discriminately. Toxicology 2016, 357–358, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Papait, R.; Serio, S.; Condorelli, G. Role of the epigenome in heart failure. Physiol. Rev. 2020, 100, 1753–1777. [Google Scholar] [CrossRef]
- Van Weerd, J.H.; Koshiba-Takeuchi, K.; Kwon, C.; Takeuchi, J.K. Epigenetic factors and cardiac development. Cardiovasc. Res. 2011, 91, 203–211. [Google Scholar] [CrossRef]
- Basma, H.; Tatineni, S.; Dhar, K.; Qiu, F.; Rennard, S.; Lowes, B.D. Electronic cigarette extract induced toxic effect in iPS-derived cardiomyocytes. BMC Cardiovasc. Disord. 2020, 20, 357. [Google Scholar] [CrossRef] [PubMed]
- Langley-Evans, S.C. Nutritional programming of disease: Unravelling the mechanism. J. Anat. 2009, 215, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Paradis, A.N.; Gay, M.S.; Zhang, L. Binucleation of cardiomyocytes: The transition from a proliferative to a terminally differentiated state. Drug Discov. Today 2014, 19, 602–609. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol. 2014, 72, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tønnessen, T.; Kryshtal, D.O.; et al. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Cutie, S.; Payumo, A.Y.; Lunn, D.; Huang, G.N. In vitro and in vivo roles of glucocorticoid and vitamin D receptors in the control of neonatal cardiomyocyte proliferative potential. J. Mol. Cell. Cardiol. 2020, 142, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Seckl, J. Glucocorticoids, prenatal stress and the programming of disease. Horm. Behav. 2011, 59, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Krontira, A.C.; Cruceanu, C.; Binder, E.B. Glucocorticoids as Mediators of Adverse Outcomes of Prenatal Stress. Trends Neurosci. 2020, 43, 394–405. [Google Scholar] [CrossRef]
- Peng, J.; Zhou, Y.; Zhang, Z.; Wang, Z.; Gao, L.; Zhang, X.; Fang, Z.; Li, G.; Chen, H.; Yang, H.; et al. The detrimental effects of glucocorticoids exposure during pregnancy on offspring’s cardiac functions mediated by hypermethylation of bone morphogenetic protein-4. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Antolic, A.; Li, M.; Richards, E.M.; Curtis, C.W.; Wood, C.E.; Keller-Wood, M. Mechanisms of in utero cortisol effects on the newborn heart revealed by transcriptomic modeling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R323–R337. [Google Scholar] [CrossRef]
- Richards, E.M.; Wood, C.E.; Rabaglino, M.B.; Antolic, A.; Keller-Wood, M. Mechanisms for the adverse effects of late gestational increases in maternal cortisol on the heart revealed by transcriptomic analyses of the fetal septum. Physiol. Genom. 2014, 46, 547–559. [Google Scholar] [CrossRef]
- Antolic, A.; Richards, E.M.; Wood, C.E.; Keller-Wood, M. A Transcriptomic Model of Postnatal Cardiac Effects of Prenatal Maternal Cortisol Excess in Sheep. Front. Physiol. 2019, 10, 816. [Google Scholar] [CrossRef]
- Coussons-Read, M.E. Effects of prenatal stress on pregnancy and human development: Mechanisms and pathways. Obstet. Med. 2013, 6, 52–57. [Google Scholar] [CrossRef]
- van Dartel, D.A.M.; Pennings, J.L.A.; de la Fonteyne, L.J.J.; Brauers, K.J.J.; Claessen, S.; van Delft, J.H.; Kleinjans, J.C.S.; Piersma, A.H. Concentration-dependent gene expression responses to flusilazole in embryonic stem cell differentiation cultures. Toxicol. Appl. Pharmacol. 2011, 251, 110–118. [Google Scholar] [CrossRef]
- de Jong, E.; Barenys, M.; Hermsen, S.A.B.; Verhoef, A.; Ossendorp, B.C.; Bessems, J.G.M.; Piersma, A.H. Comparison of the mouse Embryonic Stem cell Test, the rat Whole Embryo Culture and the Zebrafish Embryotoxicity Test as alternative methods for developmental toxicity testing of six 1,2,4-triazoles. Toxicol. Appl. Pharmacol. 2011, 253, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yu, H.; Tan, Z.; Cai, H.; Ye, W.; Zhang, M.; Wang, P. In vitro assessing the risk of drug-induced cardiotoxicity by embryonic stem cell-based biosensor. Sens. Actuators B Chem. 2011, 155, 214–219. [Google Scholar] [CrossRef]
- Cheng, W.; Yu, Z.; Feng, L.; Wang, Y. Perfluorooctane sulfonate (PFOS) induced embryotoxicity and disruption of cardiogenesis. Toxicol. Vitr. 2013, 27, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.L.; Wang, J.D.; Xu, T.T.; Zhao, Z.; Zheng, J.J.; Ge, R.S.; Zhu, D.Y. Mitochondrial toxicity of perfluorooctane sulfonate in mouse embryonic stem cell-derived cardiomyocytes. Toxicology 2017, 382, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, I.; Fiory, F.; Perruolo, G.; Miele, C.; Beguinot, F.; Formisano, P.; Oriente, F. Potential mechanisms of bisphenol a (BPA) contributing to human disease. Int. J. Mol. Sci. 2020, 21, 5761. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Cheng, W.; Feng, Y.; Wang, W.; Liang, F.; Luo, F.; Yang, S.; Wang, Y. Combined effects of BPA and PFOS on fetal cardiac development: In vitro and in vivo experiments. Environ. Toxicol. Pharmacol. 2020, 80, 103434. [Google Scholar] [CrossRef]
- Cheng, W.; Yang, S.; Li, X.; Liang, F.; Zhou, R.; Wang, H.; Feng, Y.; Wang, Y. Low doses of BPA induced abnormal mitochondrial fission and hypertrophy in human embryonic stem cell-derived cardiomyocytes via the calcineurin-DRP1 signaling pathway: A comparison between XX and XY cardiomyocytes. Toxicol. Appl. Pharmacol. 2020, 388, 114850. [Google Scholar] [CrossRef]
- Lombó, M.; González-Rojo, S.; Fernández-Díez, C.; Herráez, M.P. Cardiogenesis impairment promoted by bisphenol A exposure is successfully counteracted by epigallocatechin gallate. Environ. Pollut. 2019, 246, 1008–1019. [Google Scholar] [CrossRef]
- Schindler, Y.L.; Garske, K.M.; Wang, J.; Firulli, B.A.; Firulli, A.B.; Poss, K.D.; Yelon, D. Hand2 elevates cardiomyocyte production during zebrafish heart development and regeneration. Development 2014, 141, 3112–3122. [Google Scholar] [CrossRef]
- Burnett, S.D.; Blanchette, A.D.; Grimm, F.A.; House, J.S.; Reif, D.M.; Wright, F.A.; Chiu, W.A.; Rusyn, I. Population-based toxicity screening in human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Appl. Pharmacol. 2019, 381, 114711. [Google Scholar] [CrossRef]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Human Studies | Animal Studies | hPSC Models | ||
|---|---|---|---|---|
| Small Animals (Rodents) | Large Animals | |||
| PROS | Data supporting DOHAD: Undernutrition [32,33,34,35,36,37] Overnutrition [38,39,40,41,42] Birth weight [23,43,44,45] Paternal contribution [1,2,46,47,48,49,50,51,52,53] ART technique [2,54,55] | Easier handling/housing and genetic manipulation [17,56] High sequence conservation with humans [57] Data supporting nutrition and pregnancy complications [58,59,60,61,62,63,64,65] | Similarities with human [56,66] Data supporting nutrition and pregnancy complications [67,68,69,70,71,72] | Unlimited supply of genetically well-defined material [73] Recapitulate embryonic development [31,74,75,76,77] Possess the complete genetic background of donor/patient [21,78] Easier to introduce and/or correct genetic variants [21] Disease modelling in human cells/tissues [21,78] |
| CONS | Necessity of long-term data, larger prospective cohorts and expensive longitudinal studies [2,79] | Physiological differences with humans [56,66] Genetic manipulation not always reflect the pathogenic mutation in human [57] | Cost and experimental duration [56] Difficult genetic manipulation [56] Ethical concerns [56,66] | Difficulty to predict in vivo readouts with only in vitro data [66] Difficulty to resemble the native tissue/multi-organ complex environment [66] Genetic instability [21] Phenotypic heterogeneity between iPSC lines [21] Incomplete maturation of iPSC-derived cells [21] |
| Model | Condition | Stimulus | Key Phenotype | Reference |
|---|---|---|---|---|
| hESC-CMs | Hyperglycemia | High-glucose exposure |
| [123] |
| hiPSC-CMs (3D microtissues) | Hyperglycemia | High-glucose exposure |
| [161] |
| hiPSC-CMs | Hyperglycemia | High-glucose exposure |
| [164] |
| hiPSC-CMs | Insulin resistance | High-palmitate exposure |
| [166] |
| hESC-CMs | Insulin resistance | TNFα and FFA exposure |
| [165] |
| hESC-CMs | HLHS | Hypoxia for 72 h |
| [174] |
| miPSC-CMs | Hypoxia | Hypoxia for 24 h |
| [173] |
| P19 ECC derived CMs | Oxidative stress | Different dose-dependent stimuli (e.g., H2O2) |
| [184] |
| hiPSC-CM | Oxidative stress | Ethanol exposure |
| [195] |
| mESC-CMs | Toxicity effects | Ethanol exposure |
| [215] |
| mESC-CMs | Oxidative stress | Cigarette smoke |
| [218] |
| hESC-CMs | Nicotine toxicity | Nicotine exposure |
| [212] |
| hiPSC-CMs | Smoke toxicity | Electronic and regular smoke extract |
| [221] |
| hIPSC-CMs | Hormones | Thyroid and glucocorticoids exposure |
| [224,225] |
| mESC-CMs | Chemicals | Flusilazole exposure |
| [234,235] |
| mESC-CMs | Chemicals | Sparfloxacin and Levofloxacin |
| [236] |
| hESC-CMs | Chemicals | Trichloroethylene and Perfluorooctane sulfonate |
| [237,238] |
| mESC-CMs | Organic compounds | BPA exposure |
| [240] |
| hESC-CMs | Organic compounds | BPA exposure |
| [241] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamberto, F.; Peral-Sanchez, I.; Muenthaisong, S.; Zana, M.; Willaime-Morawek, S.; Dinnyés, A. Environmental Alterations during Embryonic Development: Studying the Impact of Stressors on Pluripotent Stem Cell-Derived Cardiomyocytes. Genes 2021, 12, 1564. https://doi.org/10.3390/genes12101564
Lamberto F, Peral-Sanchez I, Muenthaisong S, Zana M, Willaime-Morawek S, Dinnyés A. Environmental Alterations during Embryonic Development: Studying the Impact of Stressors on Pluripotent Stem Cell-Derived Cardiomyocytes. Genes. 2021; 12(10):1564. https://doi.org/10.3390/genes12101564
Chicago/Turabian StyleLamberto, Federica, Irene Peral-Sanchez, Suchitra Muenthaisong, Melinda Zana, Sandrine Willaime-Morawek, and András Dinnyés. 2021. "Environmental Alterations during Embryonic Development: Studying the Impact of Stressors on Pluripotent Stem Cell-Derived Cardiomyocytes" Genes 12, no. 10: 1564. https://doi.org/10.3390/genes12101564
APA StyleLamberto, F., Peral-Sanchez, I., Muenthaisong, S., Zana, M., Willaime-Morawek, S., & Dinnyés, A. (2021). Environmental Alterations during Embryonic Development: Studying the Impact of Stressors on Pluripotent Stem Cell-Derived Cardiomyocytes. Genes, 12(10), 1564. https://doi.org/10.3390/genes12101564