
Comparative Genomic Analysis of Bifidobacterium bifidum Strains Isolated from Different Niches


 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain Screening
2.2. DNA Extraction, Genome Sequencing, Assembly, and Annotation
2.3. ANI Calculation, Pan-/Core Genome Analysis, Orthologous Gene Clustering, and Phylogenetic Tree Construction
2.4. Prediction of the CRISPR-Cas System, Prophages, and Bacteriocin Operons
2.5. CAZy Scanning and Determination of Carbohydrate Utilization Capacity
2.6. ARG Prediction and Antibiotic Susceptibility Testing
2.7. Data Visualization and Statistical Analysis
2.8. Data Availability
3. Results
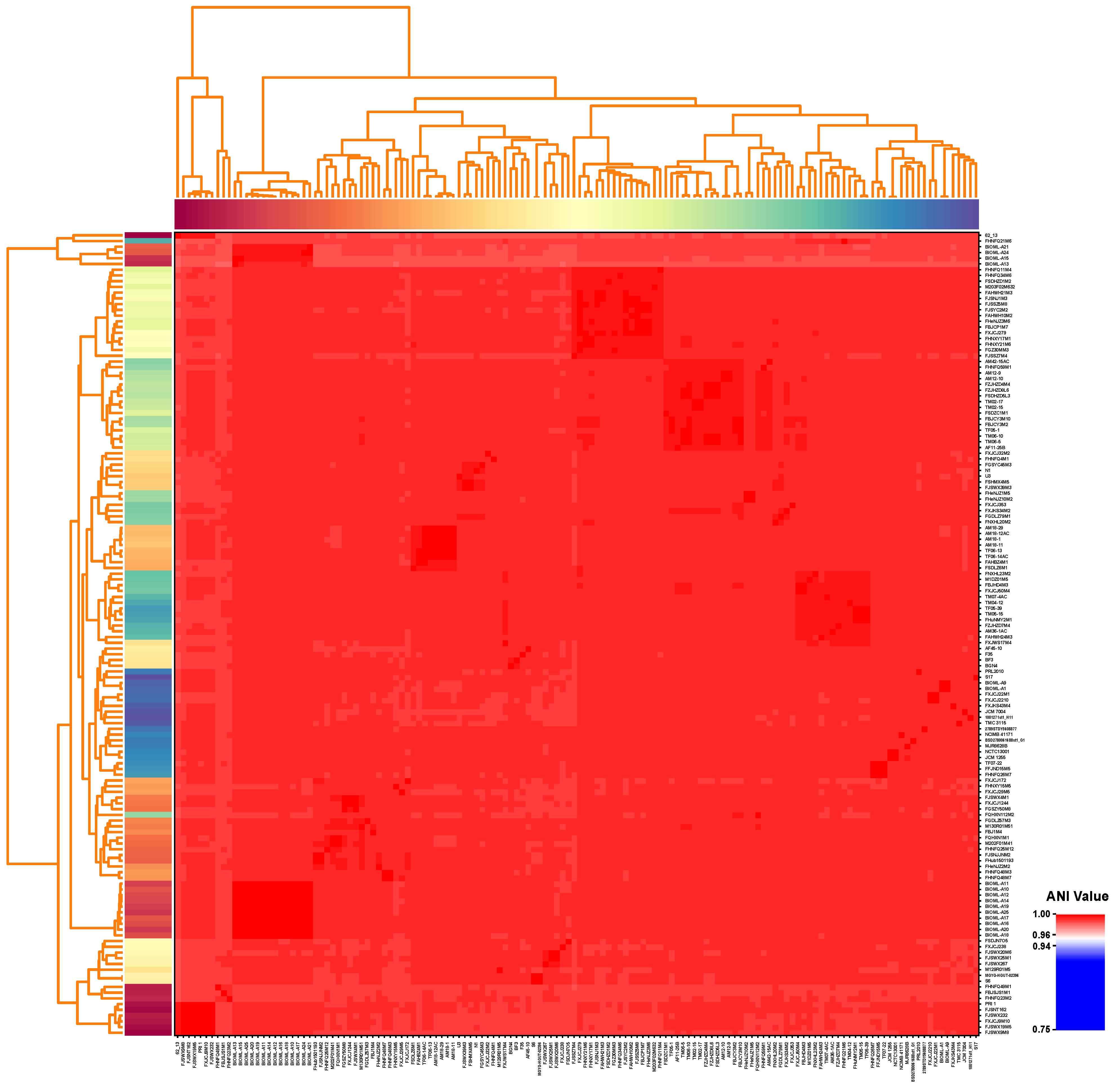
3.1. General Features and ANI Values of B. bifidum
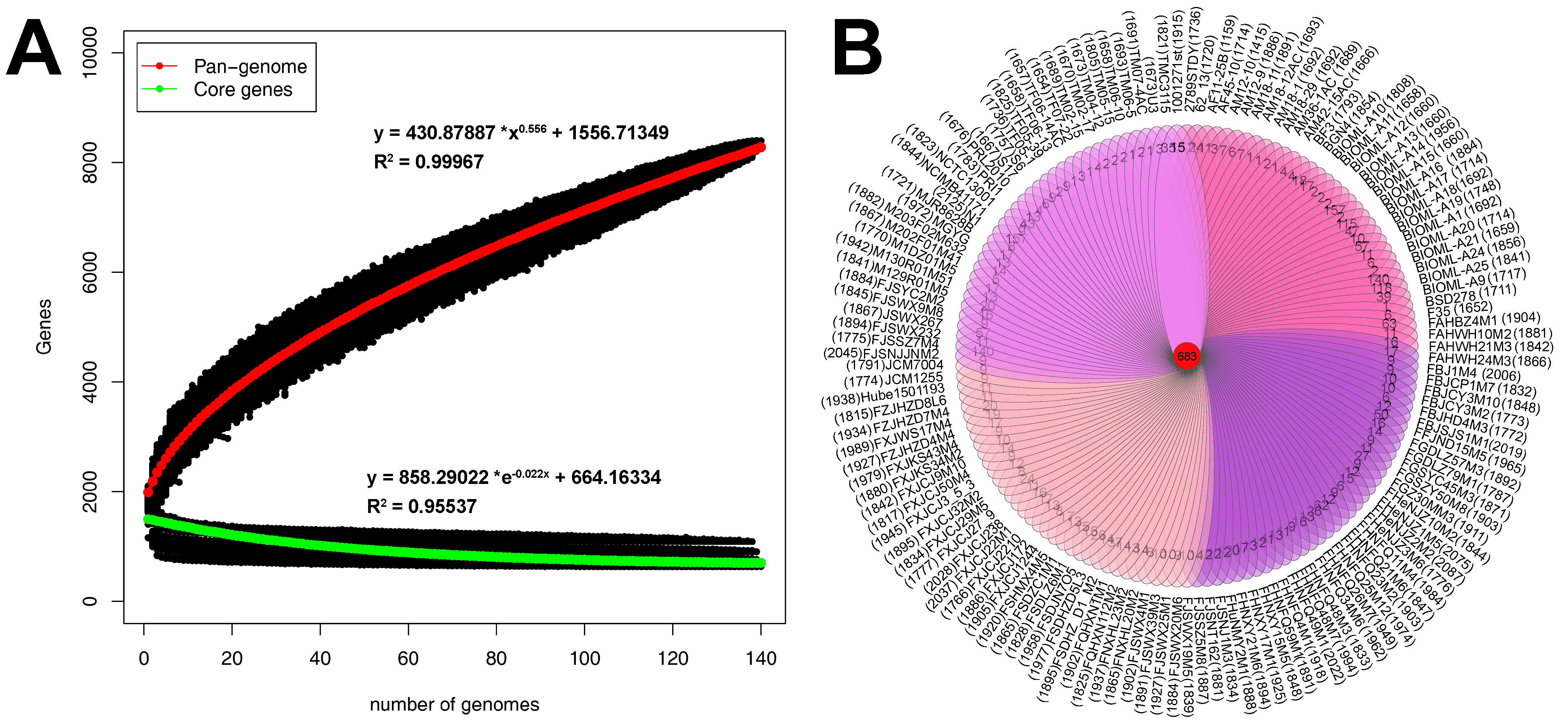
3.2. Pan-Genome and Core Genome of B. bifidum
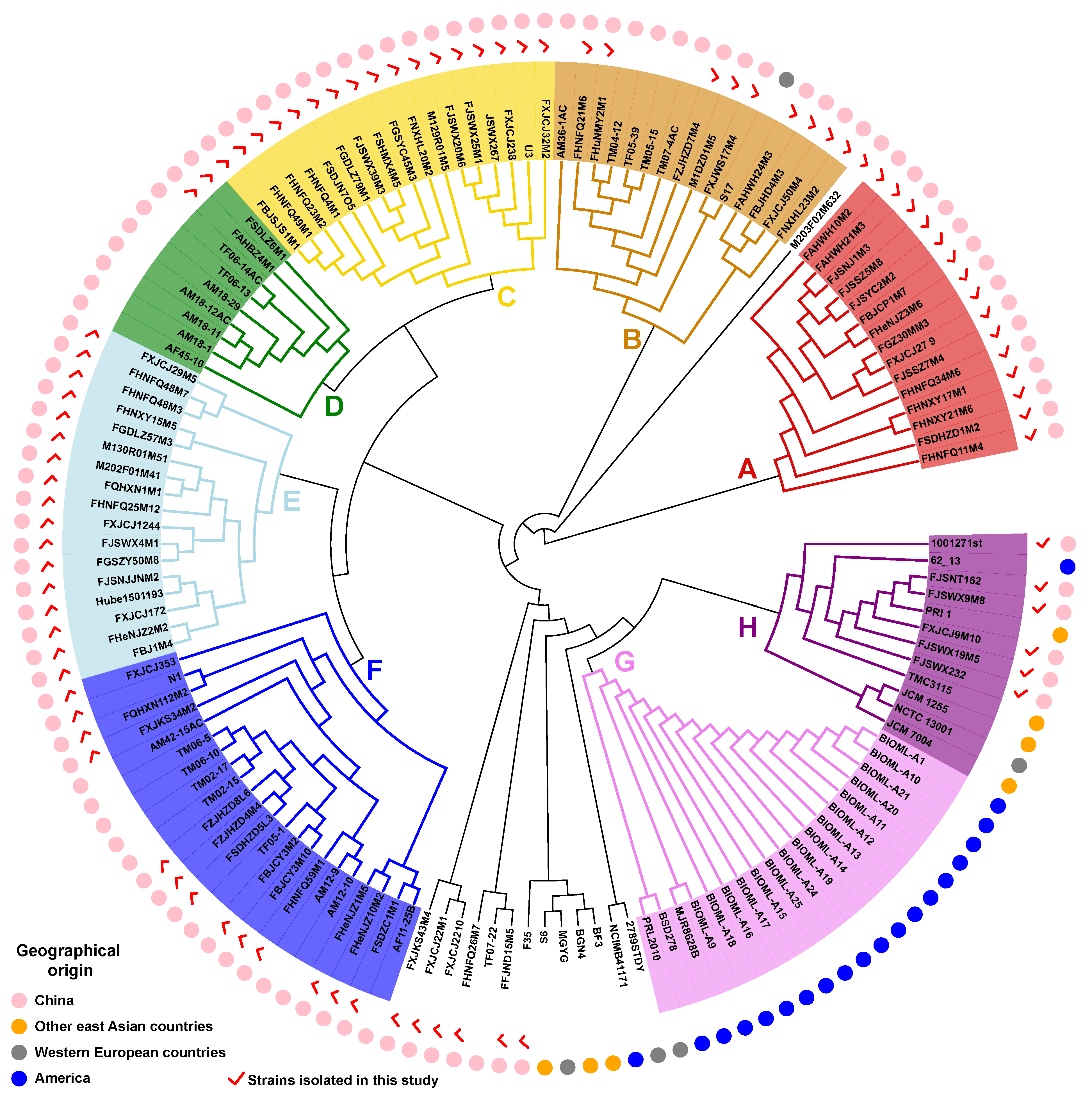
3.3. Phylogenetic Analysis of B. bifidum
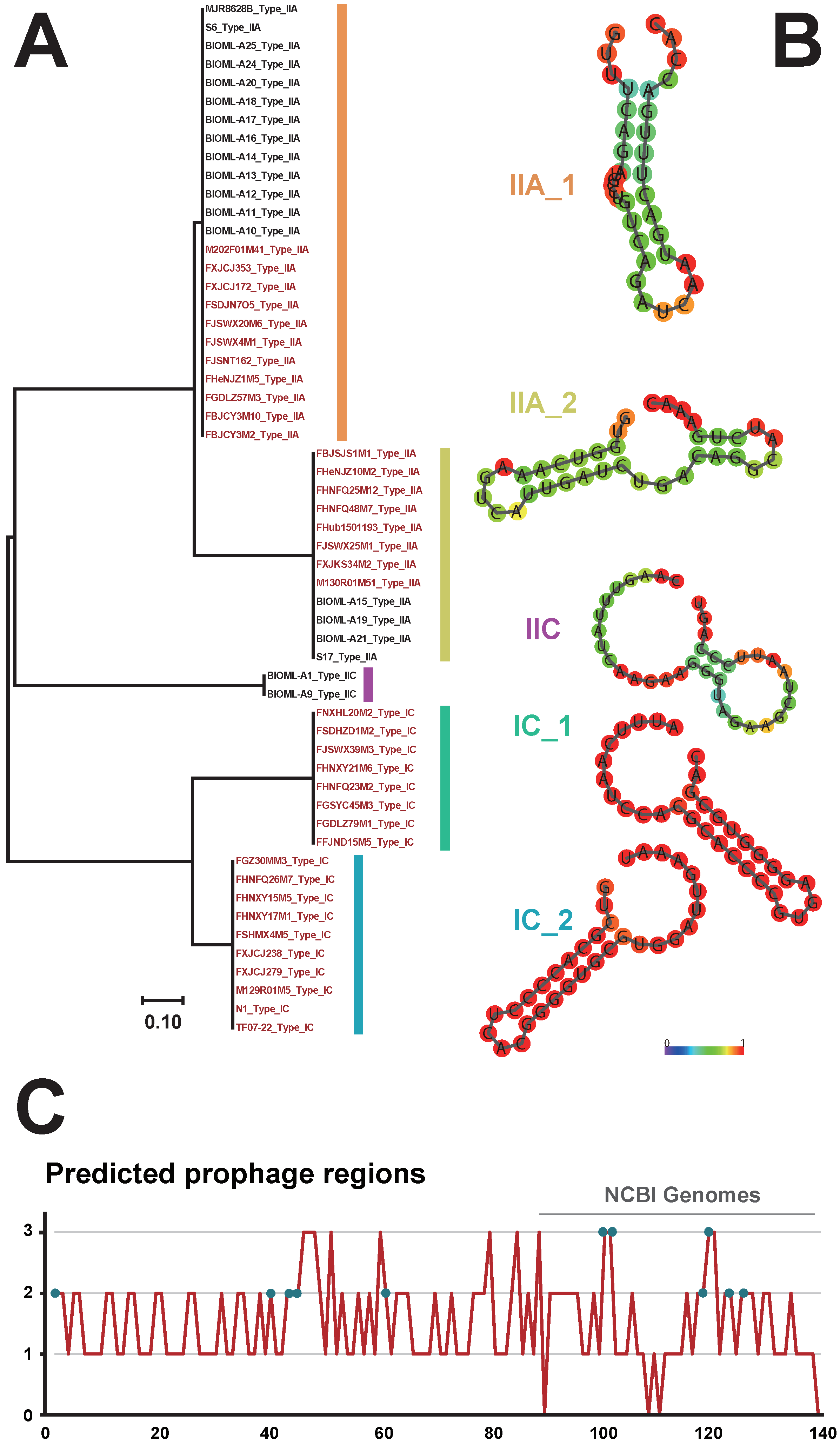
3.4. Identification of CRISPR-Cas Systems and Prophages in B. bifidum
3.5. Distribution of Bacteriocin Operons in B. bifidum
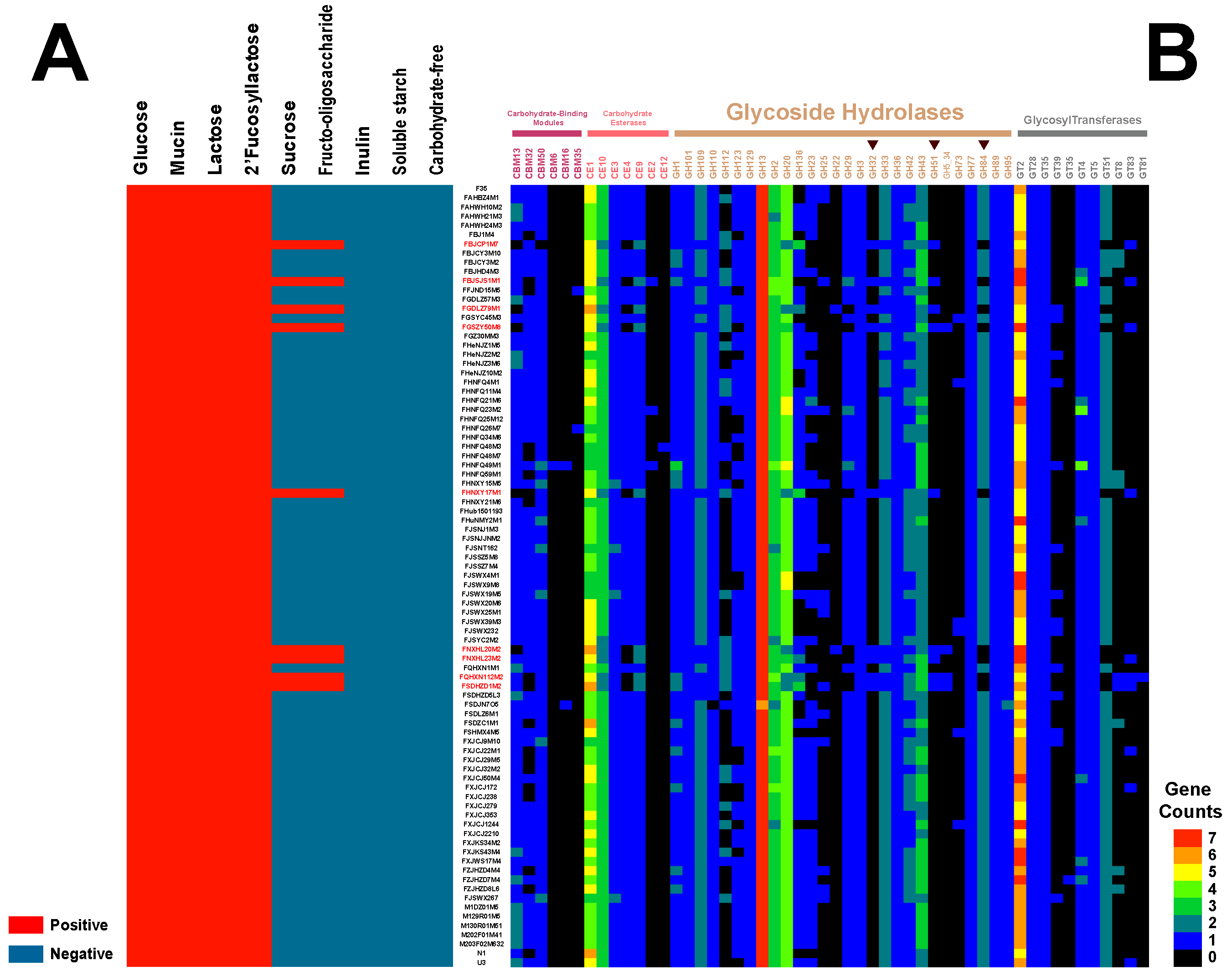
3.6. Carbohydrate Utilization Capacity and Genotype Binding Analysis
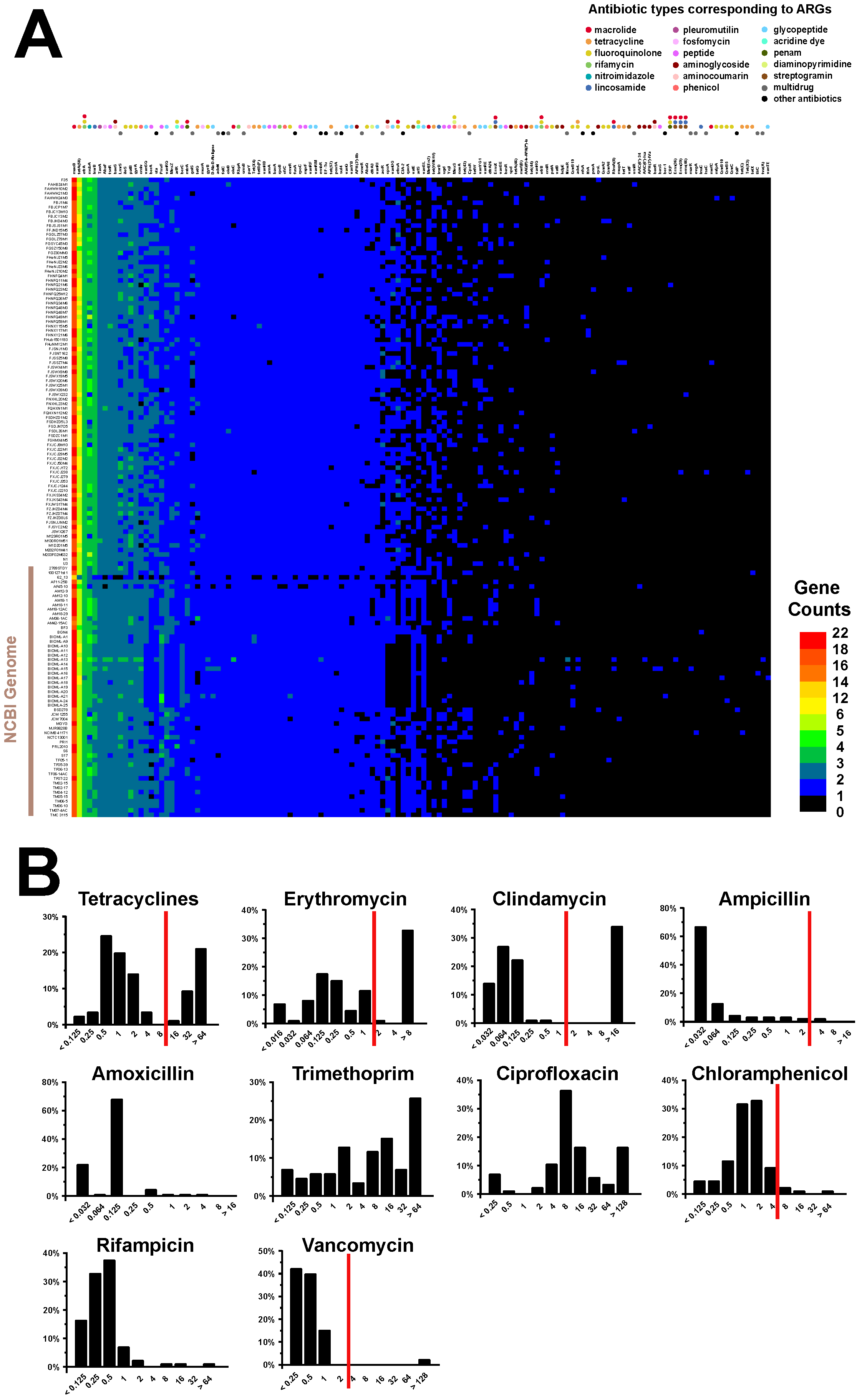
3.7. Antibiotic Resistance Genotype and Phenotype Analyses of B. bifidum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turroni, F.; Duranti, S.; Milani, C.; Lugli, G.A.; van Sinderen, D.; Ventura, M. Bifidobacterium bifidum: A key member of the early human gut microbiota. Microorganisms 2019, 7, 544. [Google Scholar] [CrossRef] [Green Version]
- Turroni, F.; Duranti, S.; Bottacini, F.; Guglielmetti, S.; Van Sinderen, D.; Ventura, M. Bifidobacterium bifidum as an example of a specialized human gut commensal. Front. Microbiol 2014, 5, 437. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Yamamoto, K. Biological analysis of the microbial metabolism of hetero-oligosaccharides in application to glycotechnology. Biosci. Biotechnol. Biochem. 2012, 76, 1815–1827. [Google Scholar] [CrossRef] [Green Version]
- Turroni, F.; Bottacini, F.; Foroni, E.; Mulder, I.; Kim, J.H.; Zomer, A.; Sanchez, B.; Bidossi, A.; Ferrarini, A.; Giubellini, V.; et al. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc. Natl. Acad. Sci. USA 2010, 107, 19514–19519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turroni, F.; Milani, C.; van Sinderen, D.; Ventura, M. Genetic strategies for mucin metabolism in Bifidobacterium bifidum PRL2010: An example of possible human-microbe co-evolution. Gut Microbes 2011, 2, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khailova, L.; Mount Patrick, S.K.; Arganbright, K.M.; Halpern, M.D.; Kinouchi, T.; Dvorak, B. Bifidobacterium bifidum reduces apoptosis in the intestinal epithelium in necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1118–G1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, K.; Wu, W.; Lv, L.; Bian, X.; Yang, L.; Wang, Q.; Li, Y.; Ye, J.; Fang, D.; et al. Administration of Bifidobacterium bifidum CGMCC 15068 modulates gut microbiota and metabolome in azoxymethane (AOM)/dextran sulphate sodium (DSS)-induced colitis-associated colon cancer (CAC) in mice. Appl. Microbiol. Biotechnol. 2020, 104, 5915–5928. [Google Scholar] [CrossRef]
- Duranti, S.; Gaiani, F.; Mancabelli, L.; Milani, C.; Grandi, A.; Bolchi, A.; Santoni, A.; Lugli, G.A.; Ferrario, C.; Mangifesta, M.; et al. Elucidating the gut microbiome of ulcerative colitis: Bifidobacteria as novel microbial biomarkers. FEMS Microbiol. Ecol. 2016, 92, fiw191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotti, I.; Turroni, F.; Piemontese, A.; Mancabelli, L.; Milani, C.; Viappiani, A.; Prevedini, G.; Sanchez, B.; Margolles, A.; Elviri, L.; et al. Evidence for cholesterol-lowering activity by Bifidobacterium bifidum PRL2010 through gut microbiota modulation. Appl. Microbiol. Biotechnol. 2015, 99, 6813–6829. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, T.; Zhang, Y.; Zheng, T.; He, Y.; He, F.; Jiang, Y. Long-term combined administration of Bifidobacterium bifidum TMC3115 and Lactobacillus plantarum 45 alleviates spatial memory impairment and gut dysbiosis in APP/PS1 mice. FEMS Microbiol. Lett. 2020, 367, fnaa048. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Lee, C.; Jeun, E.J.; Yi, J.; Kim, K.S.; Ghosh, A.; Byun, S.; Lee, C.G.; Kang, H.J.; Kim, G.C.; et al. Cell surface polysaccharides of Bifidobacterium bifidum induce the generation of Foxp3(+) regulatory T cells. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Duranti, S.; Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Sanchez, B.; Ferrario, C.; Viappiani, A.; Mangifesta, M.; Mancino, W.; et al. Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Environ. Microbiol 2015, 17, 2515–2531. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.S.; Jeong, H.; Lee, D.H.; Kwon, S.K.; Song, J.Y.; Kim, B.K.; Park, M.S.; Ji, G.E.; Oh, T.K.; Kim, J.F. Complete genome sequence of the probiotic bacterium Bifidobacterium bifidum strain BGN4. J. Bacteriol. 2012, 194, 4757–4758. [Google Scholar] [CrossRef]
- Zhurina, D.; Zomer, A.; Gleinser, M.; Brancaccio, V.F.; Auchter, M.; Waidmann, M.S.; Westermann, C.; van Sinderen, D.; Riedel, C.U. Complete genome sequence of Bifidobacterium bifidum S17. J. Bacteriol. 2011, 193, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Gueimonde, M.; Ventura, M.; Margolles, A.; Sanchez, B. Genome sequence of the immunomodulatory strain Bifidobacterium bifidum LMG 13195. J. Bacteriol. 2012, 194, 6997. [Google Scholar] [CrossRef] [Green Version]
- Andryuschenko, S.V.; Ivanova, E.V.; Perunova, N.B.; Zdvizhkova, I.A.; Bekpergenova, A.V.; Bukharin, O.V. Draft genome sequence of Bifidobacterium bifidum strain ICIS-310, isolated from the feces of a healthy 5-year-old child from Orenburg, Russia. Microbiol. Resour. Announc. 2018, 7, e01271-18. [Google Scholar] [CrossRef] [Green Version]
- Albert, K.; Rani, A.; Sela, D.A. Comparative pangenomics of the mammalian gut Commensal Bifidobacterium longum. Microorganisms 2019, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Lugli, G.A.; Mancino, W.; Milani, C.; Duranti, S.; Mancabelli, L.; Napoli, S.; Mangifesta, M.; Viappiani, A.; Anzalone, R.; Longhi, G.; et al. Dissecting the evolutionary development of the species Bifidobacterium animalis through comparative genomics analyses. Appl. Environ. Microbiol. 2019, 85, e02806-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranti, S.; Milani, C.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Sanchez, B.; Margolles, A.; et al. Evaluation of genetic diversity among strains of the human gut commensal Bifidobacterium adolescentis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Bottacini, F.; Morrissey, R.; Roberts, R.J.; James, K.; van Breen, J.; Egan, M.; Lambert, J.; van Limpt, K.; Knol, J.; Motherway, M.O.; et al. Comparative genome and methylome analysis reveals restriction/modification system diversity in the gut commensal Bifidobacterium breve. Nucleic Acids Res. 2018, 46, 1860–1877. [Google Scholar] [CrossRef]
- Toshimitsu, T.; Nakamura, M.; Ikegami, S.; Terahara, M.; Itou, H. Strain-specific identification of Bifidobacterium bifidum OLB6378 by PCR. Biosci. Biotechnol. Biochem. 2013, 77, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Deletoile, A.; Passet, V.; Aires, J.; Chambaud, I.; Butel, M.J.; Smokvina, T.; Brisse, S. Species delineation and clonal diversity in four Bifidobacterium species as revealed by multilocus sequencing. Res. Microbiol. 2010, 161, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Guo, L.; Gu, S.; Wang, O.; Zhang, R.; Peters, B.A.; Fan, G.; Liu, X.; Xu, X.; Deng, L.; et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 2020, 9, giaa094. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT: Iterative refinement and additional methods. Methods Mol. Biol. 2014, 1079, 131–146. [Google Scholar] [CrossRef]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Neron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Abby, S.S.; Neron, B.; Menager, H.; Touchon, M.; Rocha, E.P. MacSyFinder: A program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS ONE 2014, 9, e110726. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Marcu, A.; Liang, Y.; Wishart, D.S. PHAST, PHASTER and PHASTEST: Tools for finding prophage in bacterial genomes. Brief. Bioinform. 2019, 20, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- International Organization for Standardization (ISO). Milk and Milk Products: Determination of the Minimal Inhibitory Concentration (MIC) of Antibiotics Applicable to Bifidobacteria and Non-Enterococcal Lactic Acid Bacteria (LAB); ISO 10932:2010 (IDF 223:2010); ISO: Geneva, Switzerland, 2010. [Google Scholar]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A toolkit for illustrating heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Cantabrana, C.; Crawley, A.B.; Sanchez, B.; Barrangou, R. Characterization and exploitation of CRISPR loci in Bifidobacterium longum. Front. Microbiol. 2017, 8, 1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, Z.; Sadiq, F.A.; Han, X.; Zhao, J.; Zhang, H.; Ross, R.P.; Lu, W.; Chen, W. Comprehensive scanning of prophages in Lactobacillus: Distribution, diversity, antibiotic resistance genes, and linkages with CRISPR-Cas systems. mSystems 2021, 6, e01211-20. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.K.; Singh, B.; Tiwari, S.K. Comparative analysis of inhibition-based and indicator-independent colorimetric assay for screening of bacteriocin-producing lactic acid bacteria. Probiotics Antimicrob. Proteins 2019, 11, 687–695. [Google Scholar] [CrossRef]
- Yildirim, Z.; Winters, D.K.; Johnson, M.G. Purification, amino acid sequence and mode of action of bifidocin B produced by Bifidobacterium bifidum NCFB 1454. J. Appl. Microbiol. 1999, 86, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Van Heel, A.J.; Kloosterman, T.G.; Montalban-Lopez, M.; Deng, J.; Plat, A.; Baudu, B.; Hendriks, D.; Moll, G.N.; Kuipers, O.P. Discovery, production and modification of five novel Lantibiotics using the promiscuous Nisin modification machinery. ACS Synth. Biol. 2016, 5, 1146–1154. [Google Scholar] [CrossRef]
- Miescher, S.; Stierli, M.P.; Teuber, M.; Meile, L. Propionicin SM1, a bacteriocin from Propionibacterium jensenii DF1: Isolation and characterization of the protein and its gene. Syst. Appl. Microbiol. 2000, 23, 174–184. [Google Scholar] [CrossRef]
- Sheng, W.; Xu, B.; Chen, S.; Li, Y.; Liu, B.; Wang, H. Substrate tolerance of the biosynthetic enzymes of glycosylated lanthipeptide NAI-112. Org. Biomol. Chem. 2020, 18, 6095–6099. [Google Scholar] [CrossRef]
- Garg, N.; Oman, T.J.; Andrew Wang, T.S.; De Gonzalo, C.V.; Walker, S.; van der Donk, W.A. Mode of action and structure-activity relationship studies of geobacillin I. J. Antibiot. 2014, 67, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Dobson, A.; Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocin production: A probiotic trait? Appl. Environ. Microbiol. 2012, 78, 1–6. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, W.; Pei, Z.; Zang, M.; Lee, Y.-k.; Zhao, J.; Chen, W.; Wang, H.; Zhang, H. Comparative Genomic Analysis of Bifidobacterium bifidum Strains Isolated from Different Niches. Genes 2021, 12, 1504. https://doi.org/10.3390/genes12101504
Lu W, Pei Z, Zang M, Lee Y-k, Zhao J, Chen W, Wang H, Zhang H. Comparative Genomic Analysis of Bifidobacterium bifidum Strains Isolated from Different Niches. Genes. 2021; 12(10):1504. https://doi.org/10.3390/genes12101504
Chicago/Turabian StyleLu, Wenwei, Zhangming Pei, Mengning Zang, Yuan-kun Lee, Jianxin Zhao, Wei Chen, Hongchao Wang, and Hao Zhang. 2021. "Comparative Genomic Analysis of Bifidobacterium bifidum Strains Isolated from Different Niches" Genes 12, no. 10: 1504. https://doi.org/10.3390/genes12101504
APA StyleLu, W., Pei, Z., Zang, M., Lee, Y.-k., Zhao, J., Chen, W., Wang, H., & Zhang, H. (2021). Comparative Genomic Analysis of Bifidobacterium bifidum Strains Isolated from Different Niches. Genes, 12(10), 1504. https://doi.org/10.3390/genes12101504