GC and Repeats Profiling along Chromosomes—The Future of Fish Compositional Cytogenomics



Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Processing
2.2. DNA Profiling Tool
- Data download from a database such as Ensembl or NCBI, where they are accessible by the FTP. The tool saves data for every requested species into its own folder and unzips them.
- Data analysis by the sliding window approach is performed for each FASTA file separately with “DNA_puller”, a component provided on GitHub. Each window position yields the number of occurrences of each letter (i.e., ATGC), discerning the upper and the lowercase ones.
- The raw data are processed as a preparation for charts, giving GC% and the ratio between soft-masked (identified repeats) and non-soft-masked (non-repetitive DNA or not identified repeats) DNA in a CSV file for each chromosome. Such a file has three columns (index, i.e., position in DNA, GC%, and ratio) and a generally high number of rows, each of which will present a point in the chart. For instance, for a chromosome with 10 Mbp and a sliding window of size 1kbp, the result file has 10 Mbp/1 kbp = 1000 rows hence 1000 points in the chart.
- Generation of the definition files and rendering charts is a two-step process performed with the tool GNUplot, version 5.2. The former is executed with our component “gnuplot_generator”. During this step, the CSV files are sorted by the number of lines counted by the wc (‘word count’) program in Linux. Finally, the charts are rendered.
2.3. Plotting Large-Scale Profiles and Statistical Analyses
3. Results
3.1. GC-Profiles in Fish
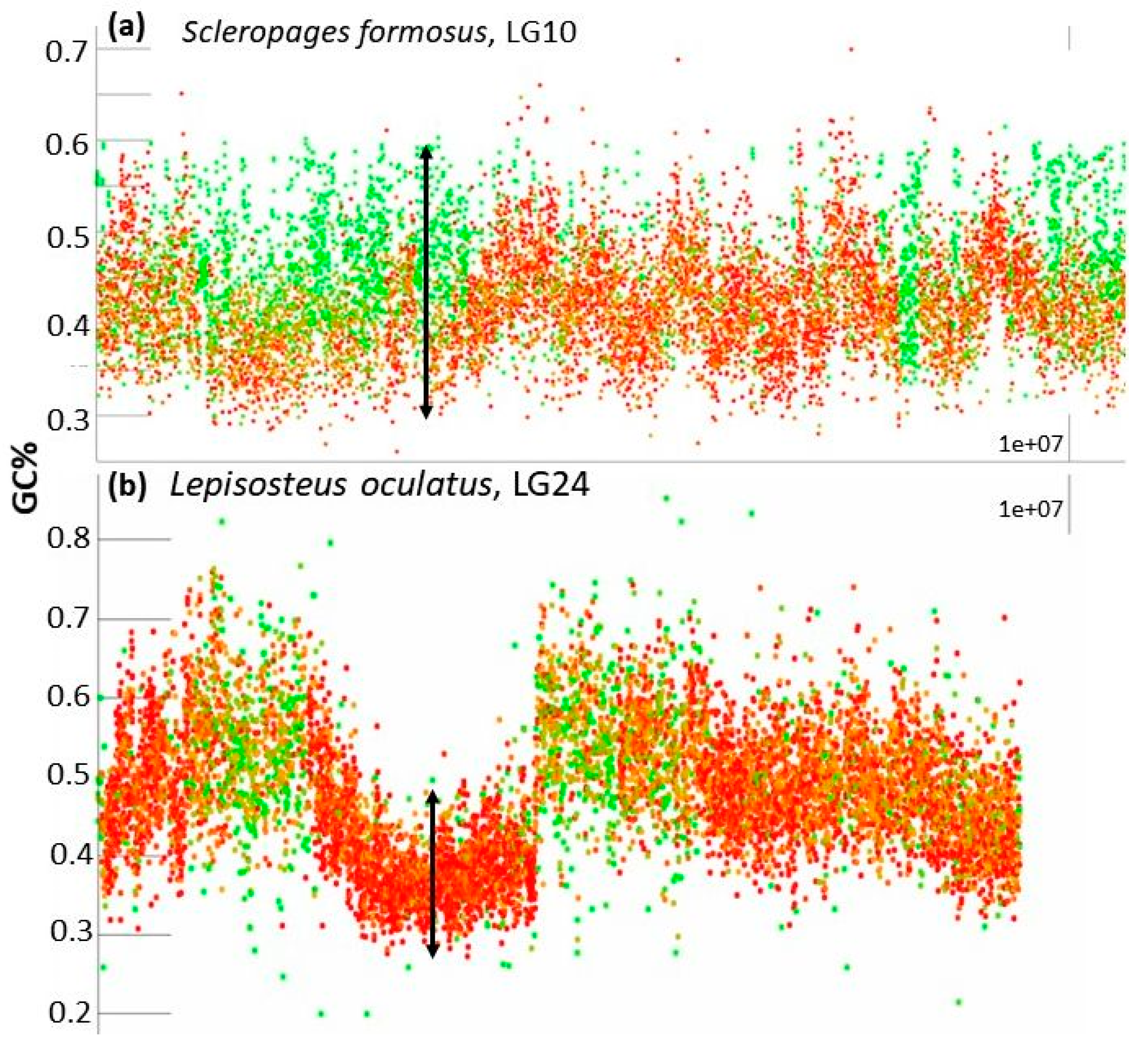
- The entire chromosome is formed by a generally flattened range of points with GC% between the minimal values around 35% and the maximal values around 55% (Oryzias latipes, Figure 2) or sometimes 30–60% (Betta splendens, Figure 2) with only rare or occasional slight departures from this pattern. Whereas some species show a narrower GC% range with almost no fluctuations/departures, e.g., in the Blunt-snouted Clingfish (Gouania willdenowi), some other species show an even broader range of GC% 30–65% with some more prominent local elevations or depletions of GC% (Scleropages formosus). Occasional slight elevations in GC% occur at the ends of chromosomes.
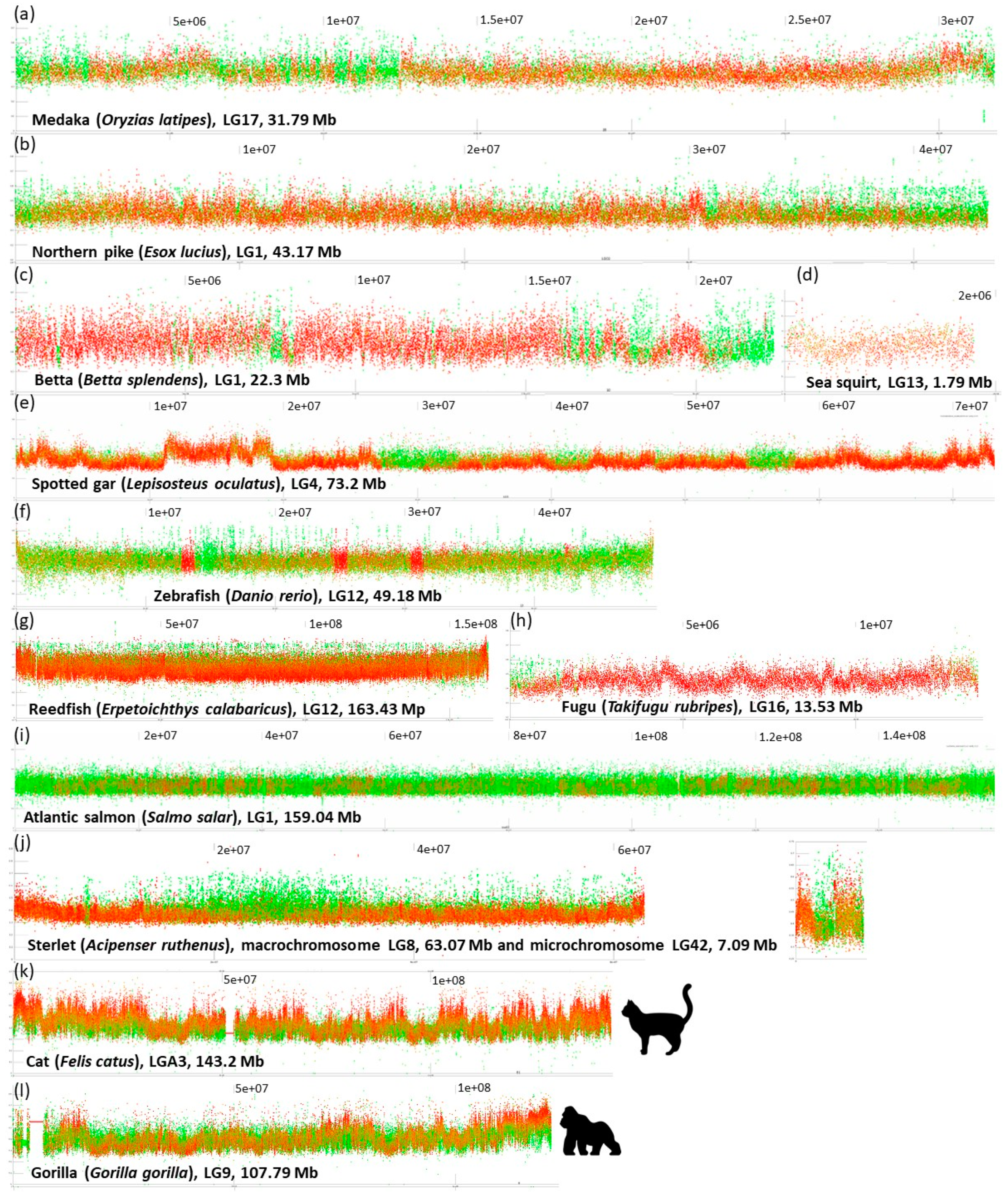
- No prominent pattern occurs in the basal chordate (tunicate) sea squirt (Ciona intestinalis). This pattern can be ascribed to an extremely low amount of DNA in the chromosomes (4.5–10 Mb). The majority of points occur in the range 30–40% of GC with only very rare and narrow peaks or isolated points reaching 50% of GC.
- So far, the only known fish species with heterogeneous AT/GC organization along LGs is the spotted gar (Lepisosteus oculatus, Figure 2). Here, a rather narrow “baseline” of densely organized points of GC% between 30–50% alters with sharp and compact peaks reaching over 60% of GC%.
- Another extreme situation exists in the reedfish (Erpetoichthys calabaricus, Figure 2) with a dense organization, however, resulting in a flat range of values between 30–55% GC. This flattened appearance can be ascribed to the exceptionally large size of chromosomes (88.37–350.1 Mb) that are even larger than mammalian chromosomes (gorilla 32.72–219.76 Mb).
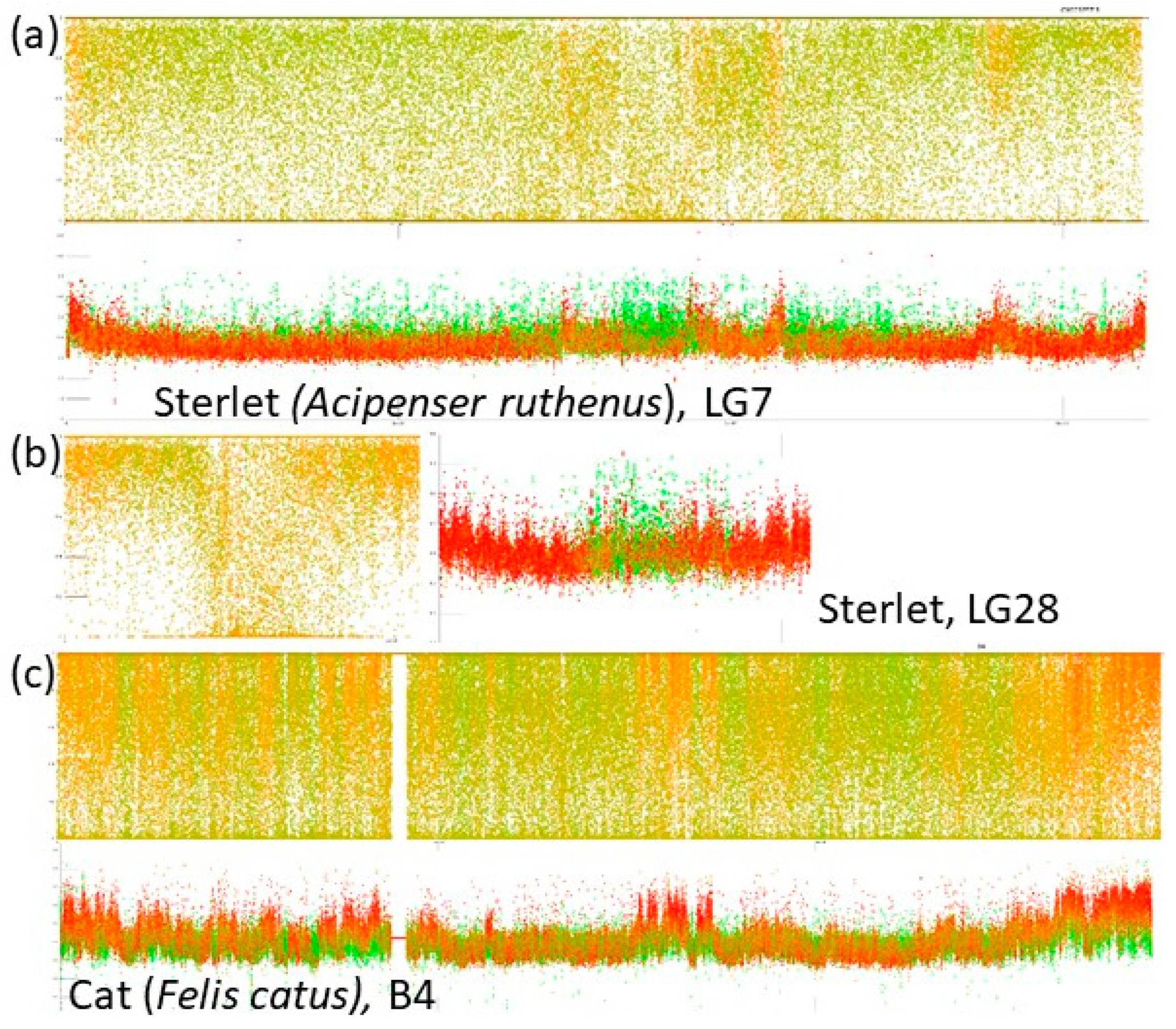
- A combination of a flattened range of GC% values in large(r) chromosomes (i.e., macrochromosomes) and more or less clear GC% elevations in smaller chromosomes (i.e., microchromosomes) exists in the sterlet (Acipenser ruthenus, Figure 2j) and all three chondrichthyan species analysed (Amblyraja radiata, Chiloscyllium plagiosum, and Pristis pectinata). Here, with the decreasing chromosome size, elevations in GC% firstly appear at the ends of chromosomes. In smaller chromosomes, internal GC% fluctuations occur.
3.2. Repeats Content and Organization in Fish
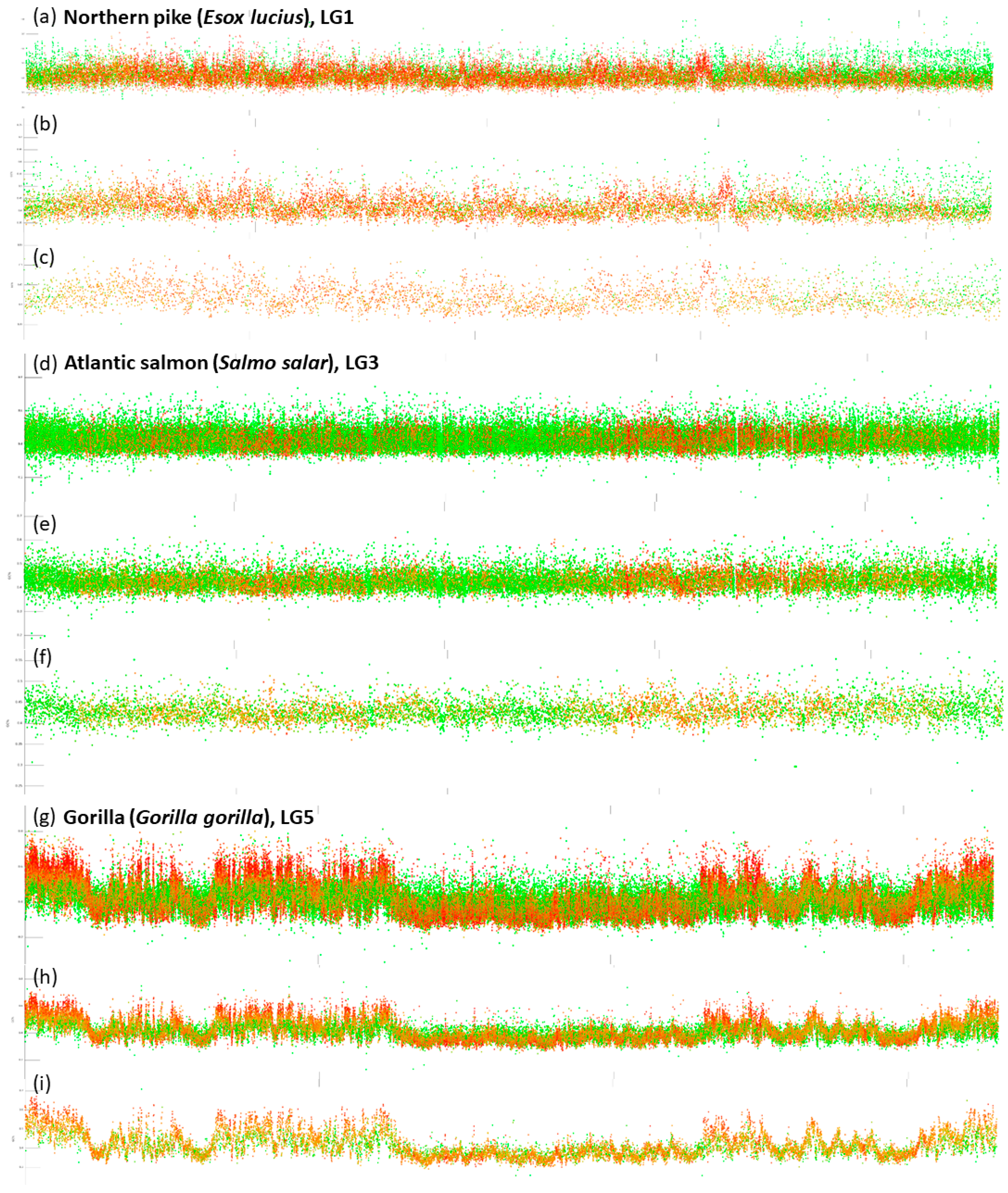
- Blocks of repeats prevailing over the non-repetitive DNA at both ends of chromosomes. This pattern is particularly prominent in species with all acrocentric chromosomes (e.g., Esox lucius (Figure 2; [44]), Oreochromis niloticus [45], Sparus aurata, etc.). The size of these blocks of repeats varies within and among species.
- Interstitial, clearly delineated small blocks of almost exclusively repetitive DNA. (e.g., Betta splendens, Figure 2, Ictalurus punctatus, Scleropages formosus, Oryzias latipes).
- Dispersed and intermingled repeats occurring mostly in fish species with larg(er) genomes (e.g., Danio rerio, Astyanax mexicanus, and pseudotetraploid salmonids Oncorhynchus mykiss and Salmo salar). Here, either completely green or orange regions of varying size are interrupted with small blocks of non-repetitive DNA.
- Limited extent of repeats proportion caused by reduced genome size through repeats elimination (Tetraodon nigroviridis, Takifugu rubripes, Figure 2, Gasterosteus aculeatus) or through insufficient repeat-masking (Oryzias javanicus, Scophthalmus maximus, etc.).
3.3. GC- and Repeat-Content in Selected Mammals and Comparison with Fish
3.4. Different Sliding Window Sizes in Fish and Mammals
3.5. Relationship between GC% and Repeats Percentage in Fishes and Mammals
3.6. Functionality of the Tool
4. Discussion
4.1. Technical Requirements and Limitations
4.2. GC- and Repeats-Profiling and Chromosome Banding in Fish
4.3. Towards Understanding the AT/GC Homogeneity of Fish Genomes
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Order | 2n 1 | Genome Size (pg) 2 | GC% |
|---|---|---|---|---|
| Acipenser ruthenus | Acipenseriformes | 120 | 1.8 | 39.8 |
| Amblyraja radiata | Rajiformes | 98 | 2.17 | 44.6 |
| Amphiprion percula | Ovalentaria | 48 | 0.9 | 39.5 |
| Astatotilapia calliptera | Cichliformes | 46 | NA | 41.1 |
| Astyanax mexicanus | Characiformes | 50 | ~1.5 | 38.4 |
| Betta splendens | Anabantiformes | 42 | 0.64 | 45.2 |
| Carassius auratus | Cypriniformes | 50 | 1.8 | 37.5 |
| Chiloscyllium plagiosum | Orectolobiformes | 102 | ~4.56 | 42 |
| Ciona intestinalis | Tunicata | 28 | 0.2 | 36 |
| Clupea harengus | Clupeiformes | 54 | ~0.9 | 44.2 |
| Cottoperca gobio | Perciformes | 48 | NA | 41 |
| Cynoglossus semilaevis | Pleuronectiformes | 44 | 0.62 | 41.3 |
| Cyprinus carpio | Cypriniformes | 100 | 1.8 | 37.1 |
| Danio rerio | Cypriniformes | 50 | 1.95 | 36.7 |
| Denticeps clupeoides | Clupeiformes | 40 | NA | 43.7 |
| Echeneis naucrates | Carangiformes | 48 | 0.7 | 41.4 |
| Erpetoichthys calabaricus | Polypteriformes | 36 | 4.7 | 40.1 |
| Esox lucius | Esociformes | 50 | 1.1 | 42.2 |
| Gadus morhua | Gadiformes | 46 | 0.65 | 46.3 |
| Gasterosteus aculeatus | Gasterosteiformes | 42 | 0.65 | 44.6 |
| Gouania willdenowi | Gobiesociformes | 48 | NA | 38.4 |
| Ictalurus punctatus | Siluriformes | 58 | 1 | 39.7 |
| Larimichthys crocea | Perciformes | 48 | NA | 41.4 |
| Lepisosteus oculatus | Lepisosteiformes | 58 | 1.4 | 40.1 |
| Maylandia zebra | Cichliformes | 46 | NA | 41.1 |
| Myripristis murdjan | Beryciformes | 48 | ~0.9 | 41.8 |
| Oncorhynchus mykiss | Salmoniformes | 58 | 2.7 | 43.4 |
| Oreochromis niloticus | Cichliformes | 46 | 1 | 39.9 |
| Oryzias javanicus | Beloniformes | 48 | 0.9 | 39 |
| Oryzias latipes | Beloniformes | 48 | 1 | 40.8 |
| Parambassis ranga | Ovalentaria | 48 | NA | 42.5 |
| Poecilia reticulata | Cyprinodontiformes | 46 | 0.88 | 40.3 |
| Pristis pectinata | Pristiformes | 92 | 2.8 | 42.6 |
| Salarias fasciatus | Blenniformes | 46 | 0.83 | 44.4 |
| Salmo salar | Salmoniformes | 60 | 3.15 | 43.9 |
| Scleropages formosus | Osteoglossiformes | 50 | NA | 44.1 |
| Scophthalmus maximus | Pleuronectiformes | 44 | 0.75 | 43.4 |
| Sparus aurata | Perciformes | 48 | 0.95 | 41.7 |
| Sphaeramia orbicularis | Kurtiformes | 48 | NA | 37.8 |
| Takifugu rubripes | Tetraodontiformes | 44 | 0.4 | 45.8 |
| Tetraodon nigroviridis | Tetraodontiformes | 42 | 0.43 | 46.6 |
| Xiphophorus maculatus | Cypridontiformes | 48 | 0.9 | 39.8 |
References
- Holmquist, G.P. Evolution of chromosome bands: Molecular ecology of noncoding DNA. J. Mol. Evol. 1989, 28, 469–486. [Google Scholar] [CrossRef]
- Bickmore, W.; Craig, J. Chromosome bands: Patterns in the genome; Molecular Biology Intelligence Unit. In Chapman & Hall; Landes Bioscience: New York, NY, USA; Austin, TX, USA, 1997; ISBN 978-1-57059-393-2. [Google Scholar]
- Holmquist, G.P. Chromosome bands, their chromatin flavors, and their functional features. Am. J. Hum. Genet. 1992, 51, 17–37. [Google Scholar]
- Holmquist, G.P. Chromosomal Bands and Sequence Features. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2005; ISBN 978-0-470-01617-6. [Google Scholar]
- Costantini, M.; Auletta, F.; Bernardi, G. Isochore patterns and gene distributions in fish genomes. Genomics 2007, 90, 364–371. [Google Scholar] [CrossRef]
- Melodelima, C.; Gautier, C. The GC-heterogeneity of teleost fishes. BMC Genom. 2008, 9, 632. [Google Scholar] [CrossRef] [PubMed]
- Blaxhall, P.C. Chromosome karyotyping of fish using conventional and G-banding methods. J. Fish Biol. 1983, 22, 417–424. [Google Scholar] [CrossRef]
- Schmid, M.; Guttenbach, M. Evolutionary diversity of reverse (R) fluorescent chromosome bands in vertebrates. Chromosoma 1988, 97, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Medrano, L.; Bernardi, G.; Couturier, J.; Dutrillaux, B.; Bernardi, G. Chromosome banding and genome compartmentalization in fishes. Chromosoma 1988, 96, 178–183. [Google Scholar] [CrossRef]
- Arrighi, F.E.; Hsu, T.C. Localization of heterochromatin in human chromosomes. Cytogenet. Genome Res. 1971, 10, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Howell, W.M.; Black, D.A. Controlled silver-staining of nucleolus organizer regions with a protective colloidal developer: A 1-step method. Experientia 1980, 36, 1014–1015. [Google Scholar] [CrossRef]
- Sharma, O.P.; Tripathi, N.K.; Sharma, K.K. A Review of Chromosome Banding in Fishes. In Some Aspects of Chromosome Structure and Functions; Springer: Dordrecht, The Netherlands, 2002; pp. 109–122. ISBN 978-0-7923-7057-4. [Google Scholar]
- Toledo, A.; Viegas-Péquignot, E.; Foresti, F.; Filho, T.; Dutrillaux, B. BrdU replication patterns demonstrating chromosome homoeologies in two fish species, genus. Eig. Cytogenet. Genome Res. 1988, 48, 117–120. [Google Scholar] [CrossRef]
- Lemieux, N.; Drouin, R.; Richer, C.-L. High-resolution dynamic and morphological G-bandings (GBG and GTG): A comparative study. Hum. Genet. 1990, 85, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Jankun, M.; Ocalewicz, K.; Woznicki, P. Replication C- and Fluorescent Chromosome Banding Patterns in European Whitefish, Coregonus lavaretus L. Hereditas 2004, 128, 195–199. [Google Scholar] [CrossRef]
- Fujiwara, A.; Nishida-Umehara, C.; Sakamoto, T.; Okamoto, N.; Nakayama, I.; Abe, S. Improved fish lymphocyte culture for chromosome preparation. Genetica 2001, 111, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, S.; Coluccia, E.; Cannas, R.; Cau, A.; Deiana, A.M. Replication Banding in two Mediterranean Moray eels: Chromosomal Characterization and Comparison. Genetica 2003, 119, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, S.; Deiana, A.M.; Deidda, F.; Lobina, C.; Mulas, A.; Coluccia, E. XX/XY sex chromosome system and chromosome markers in the snake eel Ophisurus serpens (Anguilliformes: Ophichtidae). Mar. Biol. Res. 2018, 14, 158–164. [Google Scholar] [CrossRef]
- Hellmer, A.; Voiculescu, I.; Schempp, W. Replication banding studies in two cyprinid fishes. Chromosoma 1991, 100, 524–531. [Google Scholar] [CrossRef]
- Daga, R.R.; Thode, G.; Amores, A. Chromosome complement, C-banding, Ag-NOR and replication banding in the zebrafish Danio rerio. Chromosome Res. 1996, 4, 29–32. [Google Scholar] [CrossRef]
- Molina, W.F.; Galetti, P.M. Early replication banding in Leporinus species (Osteichthyes, Characiformes) bearing differentiated sex chromosomes (ZW). Genetica 2007, 130, 153–160. [Google Scholar] [CrossRef]
- Zhang, Q.; Wolters, W.; Tiersch, T. Brief communication. Replication banding and sister-chromatid exchange of chromosomes of channel catfish (Ictalurus punctatus). J. Hered. 1998, 89, 348–353. [Google Scholar] [CrossRef]
- Fujiwara, A.; Fujiwara, M.; Nishida-Umehara, C.; Abe, S.; Masaoka, T. Characterization of Japanese flounder karyotype by chromosome bandings and fluorescence in situ hybridization with DNA markers. Genetica 2007, 131, 267–274. [Google Scholar] [CrossRef]
- Grützner, F.; Lütjens, G.; Rovira, C.; Barnes, D.W.; Ropers, H.; Haaf, T. Classical and molecular cytogenetics of the pufferfish (Tetraodon nigroviridis). Chromosome Res. 1999, 7, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Schemczssen-Graeff, Z.; Barbosa, P.; Castro, J.P.; da Silva, M.; de Almeida, M.C.; Moreira-Filho, O.; Artoni, R.F. Dynamics of Replication and Nuclear Localization of the B Chromosome in Kidney Tissue Cells in Astyanax scabripinnis (Teleostei: Characidae). Zebrafish 2020, 17, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. Structural and Evolutionary Genomics: Natural Selection in Genome Evolution; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Du, K.; Stöck, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 2020, 4, 841–852. [Google Scholar] [CrossRef] [PubMed]
- NCBI Genome Browser. Available online: https://www.ncbi.nlm.nih.gov/genome/browse (accessed on 30 September 2020).
- Symonová, R.; Howell, W. Vertebrate Genome Evolution in the Light of Fish Cytogenomics and rDNAomics. Genes 2018, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, D. Simultaneous fluorescent staining of R bands and specific heterochromatic regions (DA-DAPI bands) in human chromosomes. Cytogenet. Genome Res. 1980, 27, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, D. Counterstain-enhanced chromosome banding. Hum. Genet. 1981, 57, 1–14. [Google Scholar] [CrossRef]
- Wang, Y.; Minoshima, S.; Shimizu, N. Cot-1 banding of human chromosomes using fluorescence in situ hybridization with Cy3 labeling. Jpn. J. Hum. Genet. 1995, 40, 243–252. [Google Scholar] [CrossRef]
- Sumner, A.T.; Evans, H.J.; Buckland, R.A. New Technique for Distinguishing between Human Chromosomes. Nat. New Biol. 1971, 232, 31–32. [Google Scholar] [CrossRef]
- Symonová, R.; Majtánová, Z.; Arias-Rodriguez, L.; Mořkovský, L.; Kořínková, T.; Cavin, L.; Pokorná, M.J.; Doležálková, M.; Flajšhans, M.; Normandeau, E.; et al. Genome Compositional Organization in Gars Shows More Similarities to Mammals than to Other Ray-Finned Fish. J. Exp. Zool. Part B Mol. Dev. Evol. 2017, 328, 607–619. [Google Scholar] [CrossRef]
- Varadharajan, S.; Rastas, P.; Löytynoja, A.; Matschiner, M.; Calboli, F.C.F.; Guo, B.; Nederbragt, A.J.; Jakobsen, K.S.; Merilä, J. A high-quality assembly of the nine-spined stickleback (Pungitius pungitius) genome. Genome Biol. Evol. 2019, 11, 3291–3308. [Google Scholar] [CrossRef]
- Verdugo, R.A.; Orostica, K.Y. Global Visualization Tool of Genomic Data. Bioinformatics 2016, 32, 2366–2368. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org2015 (accessed on 30 September 2020).
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Gregory, T.R. Animal Genome Size Database. Available online: http://www.genomesize.com (accessed on 30 September 2020).
- Carducci, F.; Barucca, M.; Canapa, A.; Carotti, E.; Biscotti, M.A. Mobile Elements in Ray-Finned Fish Genomes. Life 2020, 10, 221. [Google Scholar] [CrossRef]
- Gao, B.; Shen, D.; Xue, S.; Chen, C.; Cui, H.; Song, C. The contribution of transposable elements to size variations between four teleost genomes. Mob. DNA 2016, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Han, M.; Peng, Z. Evolution and diversity of transposable elements in fish genomes. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Symonová, R.; Ocalewicz, K.; Kirtiklis, L.; Delmastro, G.B.; Pelikánová, Š.; Garcia, S.; Kovařík, A. Higher-order organisation of extremely amplified, potentially functional and massively methylated 5S rDNA in European pikes (Esox sp.). BMC Genom. 2017, 18, 391. [Google Scholar] [CrossRef] [PubMed]
- Supiwong, W.; Tanomtong, A.; Supanuam, P.; Seetapan, K.; Khakhong, S.; Sanoamuang, L. Chromosomal Characteristic of Nile Tilapia (Oreochromis niloticus) from Mitotic and Meiotic Cell Division by T-Lymphocyte Cell Culture. Cytologia 2013, 78, 9–14. [Google Scholar] [CrossRef][Green Version]
- Jankun, M.; Woznicki, P.; Furgala-Selezniow, G. Chromosomal evolution in the three species of Holarctic fish of the Genus Coregonus (Salmoniformes). Adv. Limnol. 2005, 60, 25–37. [Google Scholar]
- Bertollo, L.A.C.; Fontes, M.S.; Fenocchio, A.S.; Cano, J. The X1X2Y sex chromosome system in the fish Hoplias malabaricus. I. G-, C- and chromosome replication banding. Chromosome Res. 1997, 5, 493–499. [Google Scholar] [CrossRef]
- Ocalewicz, K. Identification of Early and Late Replicating Heterochromatic Regions on Platyfish (Xiphophorus maculatus) Chromosomes. Folia Biol. 2005, 53, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S. Whole-Genome Shotgun Assembly and Analysis of the Genome of Fugu rubripes. Science 2002, 297, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, O.; Aury, J.-M.; Brunet, F.; Petit, J.-L.; Stange-Thomann, N.; Mauceli, E.; Bouneau, L.; Fischer, C.; Ozouf-Costaz, C.; Bernot, A.; et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 2004, 431, 946–957. [Google Scholar] [CrossRef]
- Symonová, R.; Suh, A. Nucleotide composition of transposable elements likely contributes to AT/GC compositional homogeneity of teleost fish genomes. Mob. DNA 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Mugal, C.F.; Weber, C.C.; Ellegren, H. GC-biased gene conversion links the recombination landscape and demography to genomic base composition: GC-biased gene conversion drives genomic base composition across a wide range of species. BioEssays 2015, 37, 1317–1326. [Google Scholar] [CrossRef]
- Montoya-Burgos, J.I.; Boursot, P.; Galtier, N. Recombination explains isochores in mammalian genomes. Trends Genet. 2003, 19, 128–130. [Google Scholar] [CrossRef]
- Eyre-Walker, A. Recombination and mammalian genome evolution. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1993, 252, 237–243. [Google Scholar] [CrossRef]
- de Mendoza, A.; Hatleberg, W.L.; Pang, K.; Leininger, S.; Bogdanovic, O.; Pflueger, J.; Buckberry, S.; Technau, U.; Hejnol, A.; Adamska, M.; et al. Convergent evolution of a vertebrate-like methylome in a marine sponge. Nat. Ecol. Evol. 2019, 3, 1464–1473. [Google Scholar] [CrossRef]
- Fryxell, K.J.; Zuckerkandl, E. Cytosine Deamination Plays a Primary Role in the Evolution of Mammalian Isochores. Mol. Biol. Evol. 2000, 17, 1371–1383. [Google Scholar] [CrossRef]
- Wang, R.Y.-H.; Kuo, K.C.; Gehrke, C.W.; Huang, L.-H.; Ehrlich, M. Heat- and alkali-induced deamination of 5-methylcytosine and cytosine residues in DNA. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1982, 697, 371–377. [Google Scholar] [CrossRef]
- Mugal, C.F.; Arndt, P.F.; Holm, L.; Ellegren, H. Evolutionary Consequences of DNA Methylation on the GC Content in Vertebrate Genomes. G3 Genes Genomes Genet. 2015, 5, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. The neoselectionist theory of genome evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 8385–8390. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, R.P.; Boissinot, S. Variation in base composition underlies functional and evolutionary divergence in non-LTR retrotransposons. Mob. DNA 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.S.; Grande, T.; Wilson, M.V.H. Fishes of the World, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; ISBN 978-1-118-34233-6. [Google Scholar]
- Majtánová, Z.; Symonová, R.; Arias-Rodriguez, L.; Sallan, L.; Ráb, P. “Holostei versus Halecostomi” Problem: Insight from Cytogenetics of Ancient Nonteleost Actinopterygian Fish, Bowfin Amia calva. J. Exp. Zool. B Mol. Dev. Evol. 2017, 328, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef]
- Symonová, R.; Flajšhans, M.; Sember, A.; Havelka, M.; Gela, D.; Kořínková, T.; Rodina, M.; Rábová, M.; Ráb, P. Molecular Cytogenetics in Artificial Hybrid and Highly Polyploid Sturgeons: An Evolutionary Story Narrated by Repetitive Sequences. Cytogenet. Genome Res. 2013, 141, 153–162. [Google Scholar] [CrossRef]
- Borůvková, V.; Howell, W.M.; Matoulek, D.; Symonová, R. Quantitative approach to fish cytogenetics in the context of vertebrate genome evolution. Genes 2021. (submitted). [Google Scholar]
- de Bello Cioffi, M.; Ráb, P.; Ezaz, T.; Antonio Carlos Bertollo, L.; Lavoué, S.; Aquiar de Oliveira, E.; Sember, A.; Molina, F.; Henrique Santos de Souza, F.; Majtánová, Z.; et al. Deciphering the Evolutionary History of Arowana Fishes (Teleostei, Osteoglossiformes, Osteoglossidae): Insight from Comparative Cytogenomics. Int. J. Mol. Sci. 2019, 20, 4296. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matoulek, D.; Borůvková, V.; Ocalewicz, K.; Symonová, R. GC and Repeats Profiling along Chromosomes—The Future of Fish Compositional Cytogenomics. Genes 2021, 12, 50. https://doi.org/10.3390/genes12010050
Matoulek D, Borůvková V, Ocalewicz K, Symonová R. GC and Repeats Profiling along Chromosomes—The Future of Fish Compositional Cytogenomics. Genes. 2021; 12(1):50. https://doi.org/10.3390/genes12010050
Chicago/Turabian StyleMatoulek, Dominik, Veronika Borůvková, Konrad Ocalewicz, and Radka Symonová. 2021. "GC and Repeats Profiling along Chromosomes—The Future of Fish Compositional Cytogenomics" Genes 12, no. 1: 50. https://doi.org/10.3390/genes12010050
APA StyleMatoulek, D., Borůvková, V., Ocalewicz, K., & Symonová, R. (2021). GC and Repeats Profiling along Chromosomes—The Future of Fish Compositional Cytogenomics. Genes, 12(1), 50. https://doi.org/10.3390/genes12010050