Genetics of Peripartum Cardiomyopathy: Current Knowledge, Future Directions and Clinical Implications


 , ,
, ,
Abstract
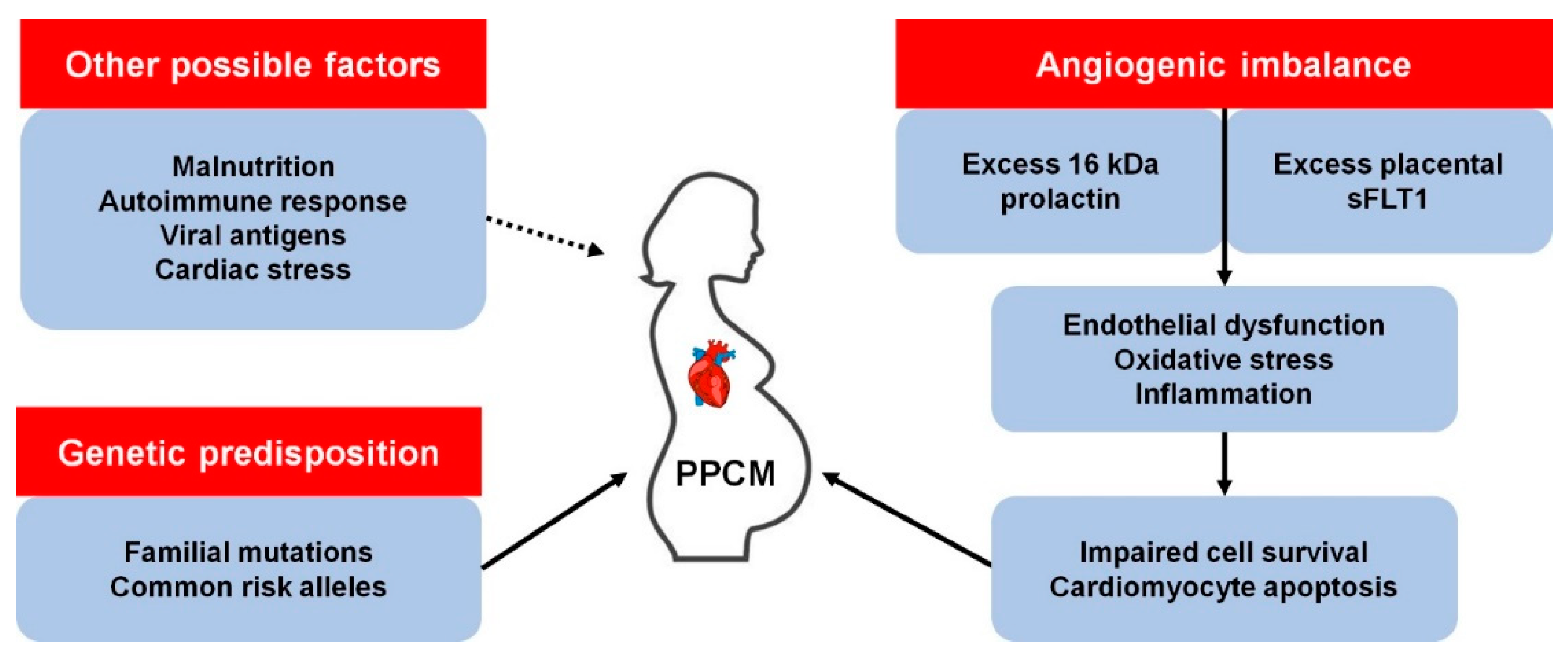
1. Introduction
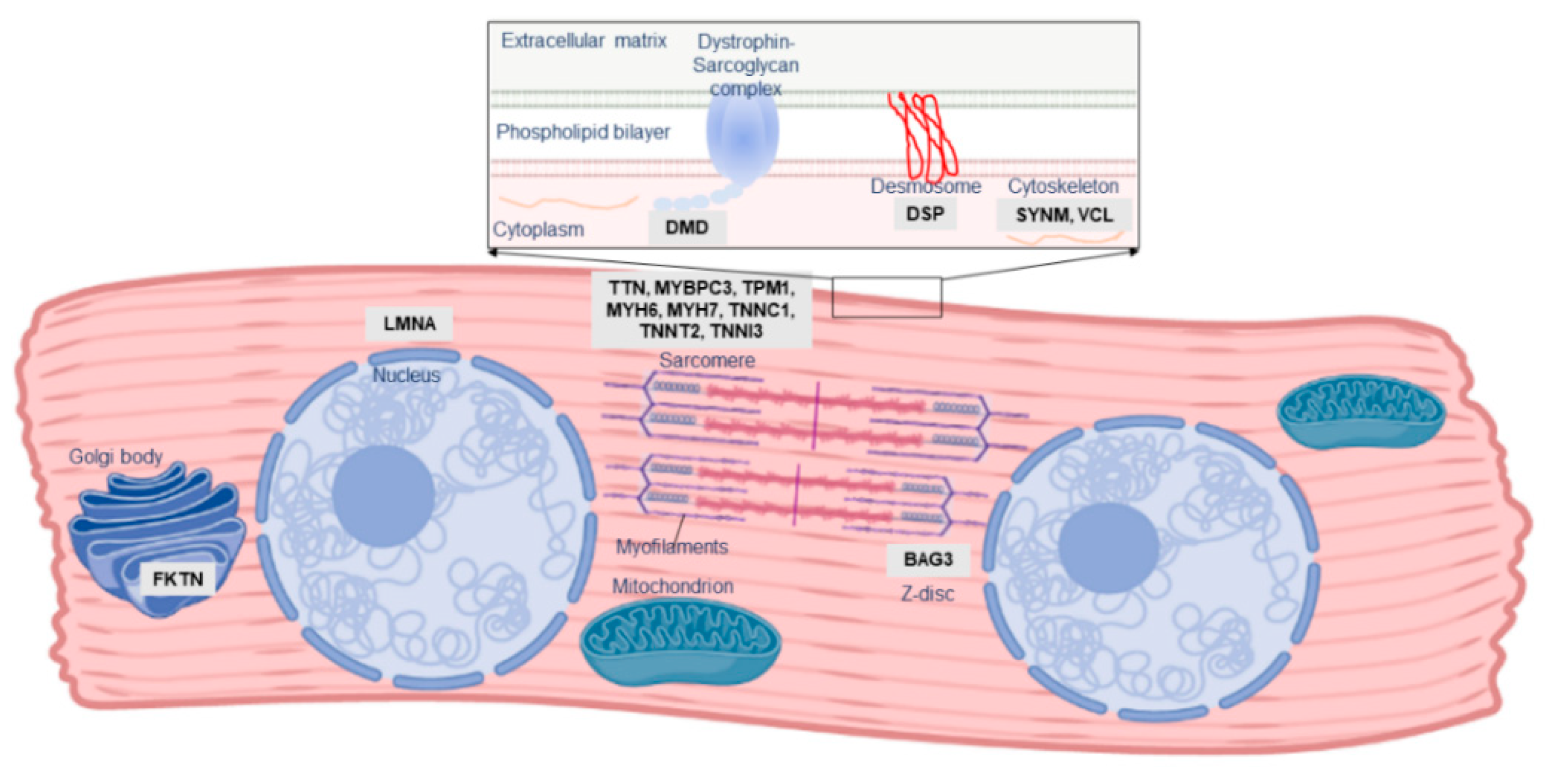
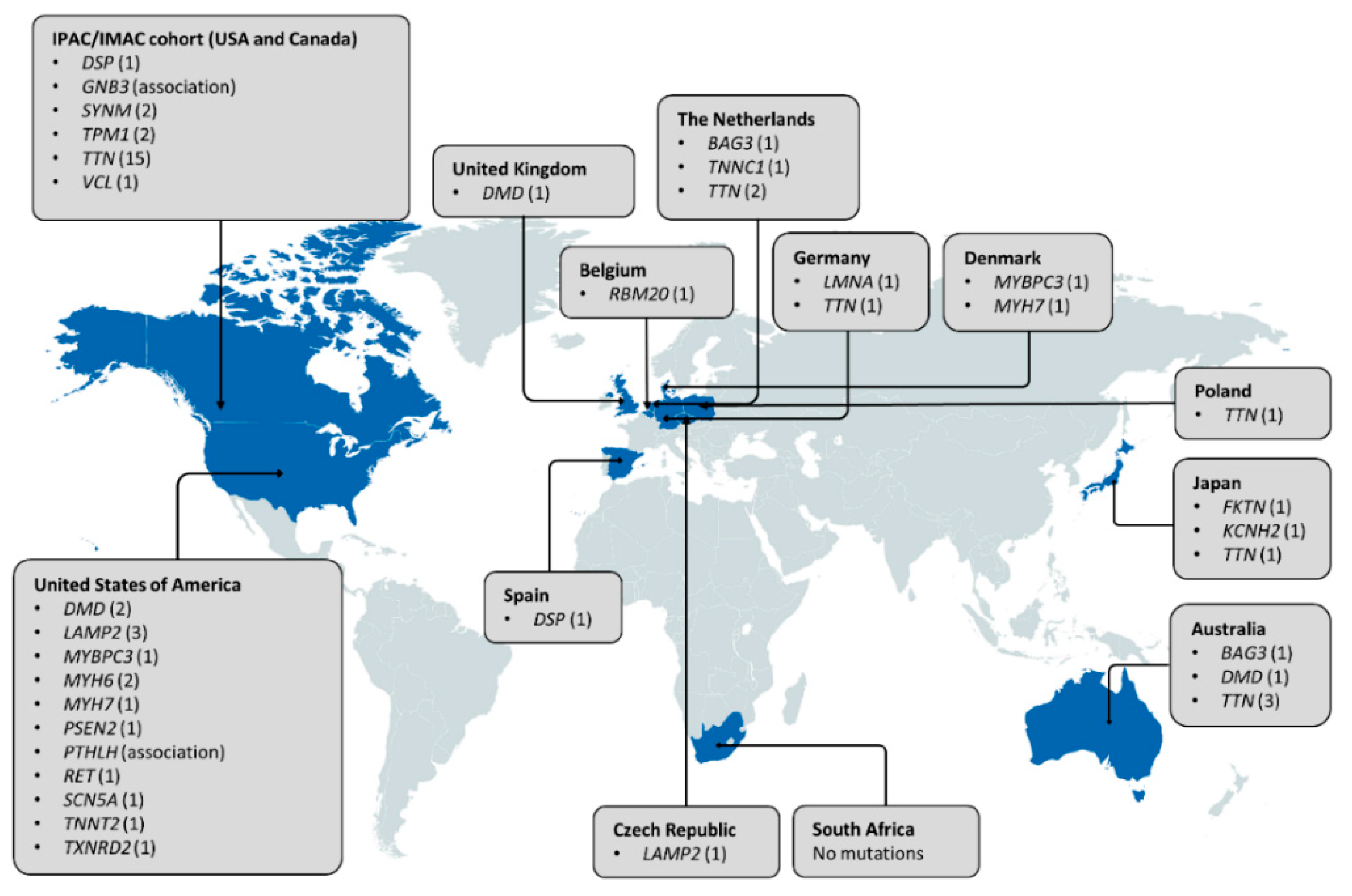
2. The Role of Familial Cardiomyopathy Genes in PPCM
3. Other Genetic Determinants of PPCM
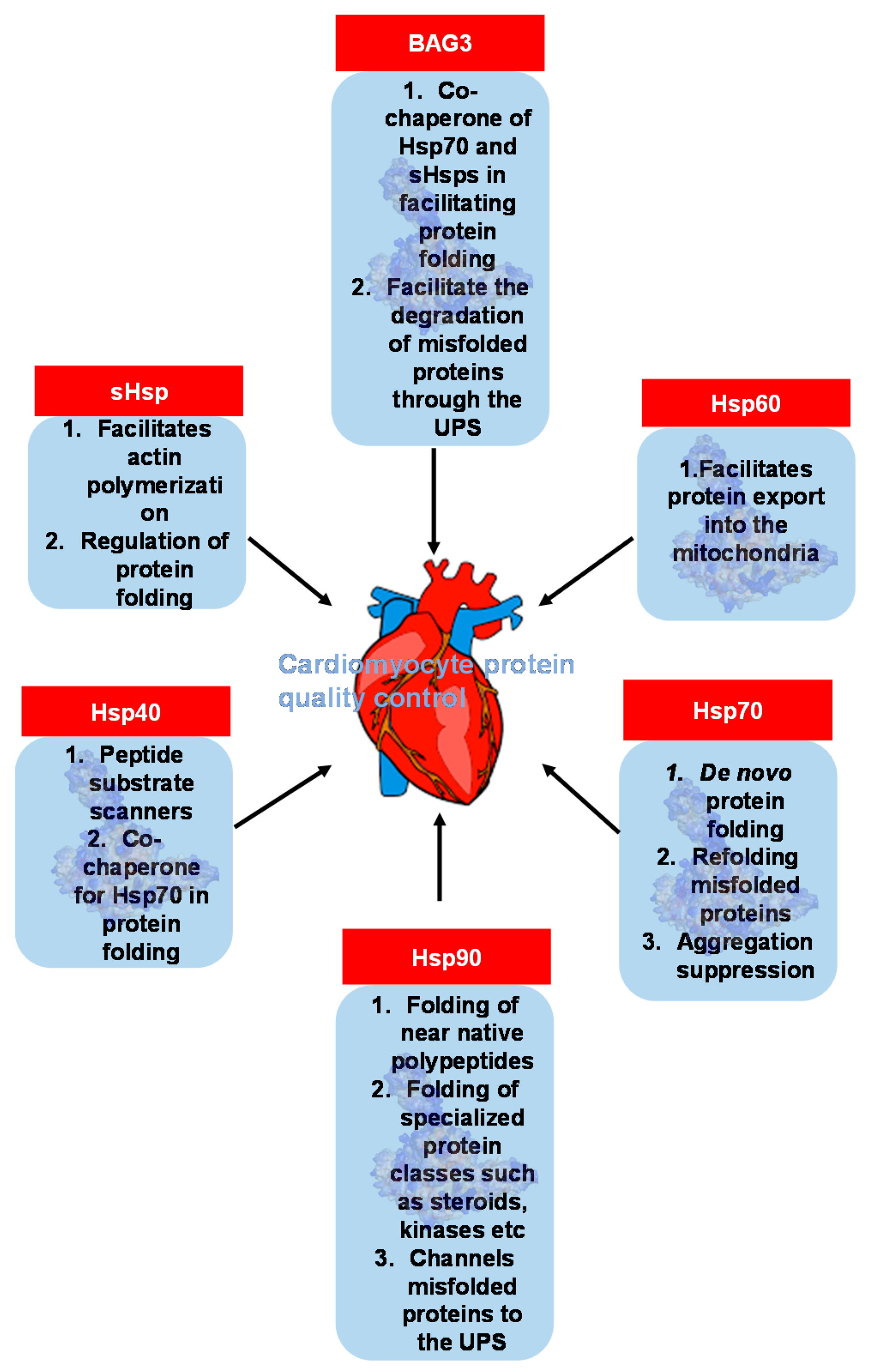
4. An Emerging Field: Heat Shock Protein and Molecular Chaperone Genes in PPCM
5. Clinical Implications
5.1. Genetic Testing May Be Indicated in PPCM Patients with Family History
5.2. Areas of Future Investigation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bauersachs, J.; Konig, T.; van der Meer, P.; Petrie, M.C.; Hilfiker-Kleiner, D.; Mbakwem, A. Pathophysiology, diagnosis and management of peripartum cardiomyopathy: A position statement from the Heart Failure Association of the European Society of Cardiology Study Group on peripartum cardiomyopathy. Eur. J. Heart Fail 2019, 21, 827–843. [Google Scholar] [CrossRef] [PubMed]
- Hoevelmann, J.; Hahnle, L.; Hahnle, J.; Sliwa, K.; Viljoen, C. Detection and management of arrhythmias in peripartum cardiomyopathy. Cardiovasc. Diagn. Ther. 2020, 10, 325–335. [Google Scholar] [CrossRef] [PubMed]
- McNamara, D.M.; Elkayam, U.; Alharethi, R.; Damp, J.; Hsich, E.; Ewald, G. Clinical outcomes for peripartum cardiomyopathy in North America: Results of the IPAC study (Investigations of Pregnancy-Associated Cardiomyopathy). J. Am. Coll. Cardiol. 2015, 66, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Moodley, J.; Naidoo, D. Peripartum cardiomyopathy: Experiences at King Edward VIII Hospital, Durban, South Africa and a review of the literature. Trop. Doct. 1995, 25, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Ersbøll, A.S.; Johansen, M.; Damm, P.; Rasmussen, S.; Vejlstrup, N.G.; Gustafsson, F. Peripartum cardiomyopathy in Denmark: A retrospective, population-based study of incidence, management and outcome. Eur. J. Heart Fail 2017, 19, 1712–1720. [Google Scholar] [CrossRef]
- Kolte, D.; Khera, S.; Aronow, W.S.; Palaniswamy, C.; Mujib, M.; Ahn, C. Temporal trends in incidence and outcomes of peripartum cardiomyopathy in the United States: A nationwide population-based study. J. Am. Heart Assoc. 2014, 3, e001056. [Google Scholar] [CrossRef]
- Sliwa, K.; Mebazaa, A.; Hilfiker-Kleiner, D.; Petrie, M.C.; Maggioni, A.P.; Laroche, C. Clinical characteristics of patients from the worldwide registry on peripartum cardiomyopathy (PPCM): EURObservational Research Programme in conjunction with the Heart Failure Association of the European Society of Cardiology Study Group on PPCM. Eur. J. Heart Fail 2017, 19, 1131–1141. [Google Scholar] [CrossRef]
- Hall, M.E.; George, E.M.; Granger, J.P. The heart during pregnancy. Rev. Esp. Cardiol. 2011, 64, 1045–1050. [Google Scholar] [CrossRef]
- Hilfiker-Kleiner, D.; Kaminski, K.; Podewski, E.; Bonda, T.; Schaefer, A.; Sliwa, K. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell 2007, 128, 589–600. [Google Scholar] [CrossRef]
- Pierce, J.A.; Price, B.O.; Joyce, J.W. Familial occurrence of postpartal heart failure. Arch Intern Med 1963, 111, 651–655. [Google Scholar] [CrossRef]
- Baruteau, A.-E.; Leurent, G.; Schleich, J.-M.; Gervais, R.; Daubert, J.-C.; Mabo, P. Can peripartum cardiomyopathy be familial? Int. J. Cardiol. 2009, 137, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Podewski, E.; Libhaber, E.; Labidi, S.; Fischer, D.; Roentgen, P. Phenotyping and outcome on contemporary management in a German cohort of patients with peripartum cardiomyopathy. Basic Res. Cardiol. 2013, 108, 366. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.; Painter, T.; Li, R.; Siegfried, J.D.; Li, D.; Norton, N. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation 2010, 121, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Ntusi, N.A.; Wonkam, A.; Shaboodien, G.; Badri, M.; Mayosi, B.M. Frequency and clinical genetics of familial dilated cardiomyopathy in Cape Town: Implications for the evaluation of patients with unexplained cardiomyopathy. S. Afr. Med. J. 2011, 1010, 394–398. [Google Scholar]
- Pearl, W. Familial occurrence of peripartum cardiomyopathy. Am. Heart J. 1995, 129, 421–492. [Google Scholar] [CrossRef]
- Rajapreyar, I.; Sinkey, R.; Pamboukian, S.V.; Tita, A. Did a shared thioredoxin-reductase gene mutation lead to maternal peripartum cardiomyopathy and fatal dilated cardiomyopathy in her son? A case report. Case Rep. Womens Health 2020, 26, e00196. [Google Scholar] [CrossRef]
- van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; van Veldhuisen, D.J.; van der Werf, R.; Jongbloed, J.D.; Paulus, W.J. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation 2010, 121, 2169–2175. [Google Scholar] [CrossRef]
- Davies, J.E.; Winokur, T.S.; Aaron, M.F.; Benza, R.L.; Foley, B.A.; Holman, W.L. Cardiomyopathy in a carrier of Duchenne’s muscular dystrophy. J. Heart Lung Transpl. 2001, 20, 781–784. [Google Scholar] [CrossRef]
- Ahmed, A.; Spinty, S.; Murday, V.; Longman, C.; Khand, A. A de-novo deletion of dystrophin provoking severe ’peri-partum cardiomyopathy: The importance of genetic testing in peripartum cardiomyopathy to uncover female carriers. Int. J. Cardiol. 2016, 203, 1084–1085. [Google Scholar] [CrossRef]
- Cheng, V.E.; Prior, D.L. Peripartum cardiomyopathy in a previously asymptomatic carrier of Duchenne muscular dystrophy. Heart Lung Circ 2013, 22, 677–681. [Google Scholar] [CrossRef]
- Gurka, J.; Piherova, L.; Majer, F.; Chaloupka, A.; Zakova, D.; Pelak, O. Danon disease is an underdiagnosed cause of advanced heart failure in young female patients: A LAMP2 flow cytometric study. ESC Heart Fail 2020. [Google Scholar] [CrossRef]
- Rowland, T.J.; Sweet, M.E.; Mestroni, L.; Taylor, M.R. Danon disease-dysregulation of autophagy in a multisystem disorder with cardiomyopathy. J. Cell Sci. 2016, 129, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Sheri, N.; Nguyen, Q.; Yokota, T. Cardiac Involvement in Dystrophin-Deficient Females: Current Understanding and Implications for the Treatment of Dystrophinopathies. Genes 2020, 11, 765. [Google Scholar] [CrossRef] [PubMed]
- Moller, D.V.; Andersen, P.S.; Hedley, P.; Ersboll, M.K.; Bundgaard, H.; Moolman-Smook, J. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur. J. Hum. Genet. 2009, 17, 1241–1249. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef]
- Tharp, C.A.; Haywood, M.E.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. The giant protein Titin’s role in cardiomyopathy: Genetic, transcriptional, and post-translational modifications of TTN and their contribution to cardiac disease. Front. Physiol. 2019, 10, 1436. [Google Scholar] [CrossRef]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200. [Google Scholar] [CrossRef]
- Myers, V.D.; Tomar, D.; Madesh, M.; Wang, J.; Song, J.; Zhang, X.-Q. Haplo-insufficiency of Bcl2-associated Athanogene 3 in mice results in progressive left ventricular dysfunction, β-adrenergic insensitivity and increased apoptosis. J. Cell Physiol. 2018, 233, 6319–6326. [Google Scholar] [CrossRef] [PubMed]
- van Spaendonck-Zwarts, K.Y.; Posafalvi, A.; van den Berg, M.P.; Hilfiker-Kleiner, D.; Bollen, I.A.; Sliwa, K. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur. Heart. J. 2014, 35, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N. Engl. J. Med. 2016, 374, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Minoche, A.E.; Horvat, C.; Johnson, R.; Gayevskiy, V.; Morton, S.U.; Drew, A.P. Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet. Med. 2019, 21, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Kryczka, K.E.; Dzielińska, Z.; Franaszczyk, M.; Wojtkowska, I.; Henzel, J.; Śpiewak, M. Severe course of peripartum cardiomyopathy and subsequent recovery in a patient with a novel TTN gene-truncating mutation. Am. J. Case Rep. 2018, 19, 820–824. [Google Scholar] [CrossRef]
- Nishimoto, O.; Matsuda, M.; Nakamoto, K.; Nishiyama, H.; Kuraoka, K.; Taniyama, K. Peripartum cardiomyopathy presenting with syncope due to Torsades de pointes: A case of long QT syndrome with a novel KCNH2 mutation. Intern. Med. 2012, 51, 461–464. [Google Scholar] [CrossRef][Green Version]
- Kim, J.; Reutrakul, S.; Davis, D.B.; Kaplan, E.L.; Refetoff, S. Multiple endocrine neoplasia 2A syndrome presenting as peripartum cardiomyopathy due to catecholamine excess. Eur. J. Endocrinol. 2004, 151, 771–777. [Google Scholar] [CrossRef]
- Glocklhofer, C.R.; Steinfurt, J.; Franke, G.; Hoppmann, A.; Glantschnig, T.; Perez-Feliz, S. A novel LMNA nonsense mutation causes two distinct phenotypes of cardiomyopathy with high risk of sudden cardiac death in a large five-generation family. Europace 2018, 20, 2003–2013. [Google Scholar] [CrossRef]
- Molina, P.; Sanz-Sanchez, J.; Fenollosa, M.; Martinez-Matilla, M.; Giner, J.; Zorio, E. Arrhythmogenic cardiomyopathy with left ventricular involvement versus ischemic heart disease: Lessons learned from the family study and the reviewed autopsy of a young male. Forensic Sci. Res. 2019, 4, 274–279. [Google Scholar] [CrossRef]
- Murakami, T.; Hayashi, Y.K.; Noguchi, S.; Ogawa, M.; Nonaka, I.; Tanabe, Y. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann. Neurol. 2006, 60, 597–602. [Google Scholar] [CrossRef]
- Robyns, T.; Willems, R.; Van Cleemput, J.; Jhangiani, S.; Muzny, D.; Gibbs, R. Whole exome sequencing in a large pedigree with DCM identifies a novel mutation in RBM20. Acta Cardiol. 2019, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2020, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Paldino, A.; De Angelis, G.; Merlo, M.; Gigli, M.; Dal Ferro, M.; Severini, G.M. Genetics of dilated cardiomyopathy: Clinical implications. Curr. Cardiol. Rep. 2018, 20, 83. [Google Scholar] [CrossRef] [PubMed]
- Horne, B.D.; Rasmusson, K.D.; Alharethi, R.; Budge, D.; Brunisholz, K.D.; Metz, T. Genome-wide significance and replication of the chromosome 12p11.22 locus near the PTHLH gene for peripartum cardiomyopathy. Circ. Cardiovasc. Genet. 2011, 4, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, R.; Hsich, E.; Damp, J.; Elkayam, U.; Kealey, A.; Ramani, G. GNB3 C825T polymorphism and myocardial recovery in peripartum cardiomyopathy: Results of the multicenter Investigations of Pregnancy-Associated Cardiomyopathy study. Circ. Heart Fail 2016, 9, e002683. [Google Scholar] [CrossRef] [PubMed]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The role of heat shock proteins and co-chaperones in heart failure. Philos. Trans. R Soc. Lond B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Myers, V.D.; McClung, J.M.; Wang, J.; Tahrir, F.G.; Gupta, M.K.; Gordon, J. The multifunctional protein BAG3: A novel therapeutic target in cardiovascular disease. JACC Basic Transl. Sci. 2018, 3, 122–131. [Google Scholar] [CrossRef]
- Lindquist, S. The heat-shock response. Ann Rev Biochem 1986, 55, 1151–1191. [Google Scholar] [CrossRef] [PubMed]
- McLendon, P.M.; Robbins, J. Proteotoxicity and cardiac dysfunction. Circ. Res. 2015, 116, 1863–1882. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Van Marion, D.M.S.; Wiersma, M.; Zhang, D.; Brundel, B.J.J.M. The protective role of small heat shock proteins in cardiac diseases: Key role in atrial fibrillation. Cell Stress Chap. 2017, 22, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Collier, M.P.; Benesch, J.L. Small heat-shock proteins and their role in mechanical stress. Cell Stress Chaper. 2020, 25, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Zhu, H.; Cai, W.F.; Wang, X.; Jiang, M.; Essandoh, K. Regulation of BECN1-mediated autophagy by HSPB6: Insights from a human HSPB6(S10F) mutant. Autophagy 2018, 14, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Shatov, V.M.; Gusev, N.B. Physico-chemical properties of two point mutants of small heat shock protein HspB6 (Hsp20) with abrogated cardioprotection. Biochimie 2020, 174, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Gardner, G.; Adly, G.; Jiang, M.; Cai, W.F.; Lam, C.K. A novel human S10F-Hsp20 mutation induces lethal peripartum cardiomyopathy. J. Cell Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ricke-Hoch, M.; Bultmann, I.; Stapel, B.; Condorelli, G.; Rinas, U.; Sliwa, K. Opposing roles of Akt and STAT3 in the protection of the maternal heart from peripartum stress. Cardiovasc. Res. 2014, 101, 587–596. [Google Scholar] [CrossRef]
- Matkovich, S.J.; Van Booven, D.J.; Hindes, A.; Kang, M.Y.; Druley, T.E.; Vallania, F.L. Cardiac signaling genes exhibit unexpected sequence diversity in sporadic cardiomyopathy, revealing HSPB7 polymorphisms associated with disease. J. Clin. Investig. 2010, 120, 280–289. [Google Scholar] [CrossRef]
- Stark, K.; Esslinger, U.B.; Reinhard, W.; Petrov, G.; Winkler, T.; Komajda, M. Genetic association study identifies HSPB7 as a risk gene for idiopathic dilated cardiomyopathy. PLoS Genet. 2010, 6, e1001167. [Google Scholar] [CrossRef]
- Esslinger, U.; Garnier, S.; Korniat, A.; Proust, C.; Kararigas, G.; Muller-Nurasyid, M. Exome-wide association study reveals novel susceptibility genes to sporadic dilated cardiomyopathy. PLoS ONE 2017, 12, e0172995. [Google Scholar] [CrossRef]
- Cappola, T.P.; Li, M.; He, J.; Ky, B.; Gilmore, J.; Qu, L. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circ. Cardiovasc. Genet. 2010, 3, 147–154. [Google Scholar] [CrossRef]
- Garnier, S.; Hengstenberg, C.; Lamblin, N.; Dubourg, O.; De Groote, P.; Fauchier, L. Involvement of BAG3 and HSPB7 loci in various etiologies of systolic heart failure: Results of a European collaboration assembling more than 2000 patients. Int. J. Cardiol. 2015, 189, 105–107. [Google Scholar] [CrossRef]
- Chen, F.F.; Xia, Y.L.; Xu, C.Q.; Li, S.S.; Zhao, Y.Y.; Wang, X.J. Common variant rs7597774 in ADD2 is associated with dilated cardiomyopathy in Chinese Han population. Int. J. Clin. Exp. Med. 2015, 8, 1188–1196. [Google Scholar] [PubMed]
- Li, X.; Luo, R.; Mo, X.; Jiang, R.; Kong, H.; Hua, W. Polymorphism of ZBTB17 gene is associated with idiopathic dilated cardiomyopathy: A case control study in a Han Chinese population. Eur. J. Med. Res. 2013, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Sanbe, A. Molecular mechanisms of α-crystallinopathy and its therapeutic strategy. Biol. Pharm. Bull. 2011, 34, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A. The novel alphaB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef] [PubMed]
- van der Smagt, J.J.; Vink, A.; Kirkels, J.H.; Nelen, M.; ter Heide, H.; Molenschot, M.M. Congenital posterior pole cataract and adult onset dilating cardiomyopathy: Expanding the phenotype of alphaB-crystallinopathies. Clin. Genet. 2014, 85, 381–385. [Google Scholar] [CrossRef]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prévost, M.C.; Faure, A. A missense mutation in the alphaB-crystallin chaperone gene causes desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef]
- Inagaki, N.; Hayashi, T.; Arimura, T.; Koga, Y.; Takahashi, M.; Shibata, H. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 342, 379–386. [Google Scholar] [CrossRef]
- Pilotto, A.; Marziliano, N.; Pasotti, M.; Grasso, M.; Costante, A.M.; Arbustini, E. alphaB-crystallin mutation in dilated cardiomyopathies: Low prevalence in a consecutive series of 200 unrelated probands. Biochem. Biophys. Res. Commun. 2006, 346, 1115–1117. [Google Scholar] [CrossRef]
- Enomoto, H.; Mittal, N.; Inomata, T.; Arimura, T.; Izumi, T.; Kimura, A. Dilated Cardiomyopathy (DCM)-linked Heat shock protein Family D Member 1 (HSPD1) mutations cause upregulation of ROS and autophagy through mitochondrial dysfunction. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Fan, F.; Duan, Y.; Yang, F.; Trexler, C.; Wang, H.; Huang, L. Deletion of heat shock protein 60 in adult mouse cardiomyocytes perturbs mitochondrial protein homeostasis and causes heart failure. Cell Death Differ. 2020, 27, 587–600. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C. Current diagnostic and treatment strategies for specific dilated cardiomyopathies: A scientific statement from the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011, 124, e783–e831. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Bohm, M. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm. 2019, 16, e373–e407. [Google Scholar] [CrossRef]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef]
- Vears, D.F.; Sénécal, K.; Borry, P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur. J. Med. Genet. 2017, 60, 553–558. [Google Scholar] [CrossRef]
- Catchpool, M.; Ramchand, J.; Martyn, M.; Hare, D.L.; James, P.A.; Trainer, A.H. A cost-effectiveness model of genetic testing and periodical clinical screening for the evaluation of families with dilated cardiomyopathy. Genet. Med. 2019, 21, 2815–2822. [Google Scholar] [CrossRef]
- Ingles, J.; McGaughran, J.; Scuffham, P.A.; Atherton, J.; Semsarian, C. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart 2012, 98, 625–630. [Google Scholar] [CrossRef]
- Van Tintelen, J.P.; Pieper, P.G.; Van Spaendonck-Zwarts, K.Y.; Van Den Berg, M.P. Pregnancy, cardiomyopathies, and genetics. Cardiovasc. Res. 2014, 101, 571–578. [Google Scholar] [CrossRef]
- Schaufelberger, M. Cardiomyopathy and pregnancy. Heart 2019, 105, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, K.; Petrie, M.C.; van der Meer, P.; Mebazaa, A.; Hilfiker-Kleiner, D.; Jackson, A.M. Clinical presentation, management, and 6-month outcomes in women with peripartum cardiomyopathy: An ESC EORP registry. Eur Heart J 2020, 41, 3737–3797. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Karaye, K.M.; Ishaq, N.A.; Sa’idu, H.; Balarabe, S.A.; Talle, M.A.; Isa, M.S. Incidence, clinical characteristics, and risk factors of peripartum cardiomyopathy in Nigeria: Results from the PEACE Registry. ESC Heart Fail 2020, 7, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Azibani, F.; Pfeffer, T.J.; Ricke-Hoch, M.; Dowling, W.; Pietzsch, S.; Briton, O. Outcome in German and South African peripartum cardiomyopathy cohorts associates with medical therapy and fibrosis markers. ESC Heart Fail 2020, 7, 512–522. [Google Scholar] [CrossRef]
- Shaboodien, G.; Spracklen, T.F.; Kamuli, S.; Ndibangwi, P.; Van Niekerk, C.; Ntusi, N.A.B. Genetics of inherited cardiomyopathies in Africa. Cardiovasc. Diagn. Ther. 2020, 10, 262–278. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Molecular Function | Mutation Types in PPCM | Other Associated Disorders |
|---|---|---|---|
| BAG3 | Co-chaperone, Z disk | Truncating | DCM, MFM |
| DMD | Sarcolemma, structure | Truncating | DCM, MD |
| DSP | Desmosome, cell–cell adhesion | Truncating | ACM, DCM, keratodermas |
| FKTN | May process dystrophin | Truncating/missense | DCM, MD |
| GNB3 | G protein subunit | Association with outcome | Hypertension, night blindness |
| KCNH2 | K+ channel, cardiac conduction | Truncating | Long QT syndrome |
| LAMP2 | Lysosome, autophagy | Truncating/missense | Danon disease, DCM, HCM |
| LMNA | Nuclear lamina, structure | Truncating | DCM, MD |
| MYBPC3 | Sarcomere, cardiac contraction | Missense | DCM, HCM, LVNC |
| MYH6 | Sarcomere, cardiac contraction | Truncating/missense | CHD, DCM, HCM |
| MYH7 | Sarcomere, cardiac contraction | Missense | DCM, HCM, LVNC, MD |
| PSEN2 | May regulate APP processing | Missense | Alzheimer’s disease, DCM |
| PTHLH | Hormone | Association with risk | Brachydactyly |
| RET | Protooncogene | Missense | Multiple endocrine neoplasia |
| SCN5A | NA+ channel, cardiac conduction | Missense | AF, DCM, Long QT syndrome, VF |
| SYNM | Cytoskeleton | Truncating | - |
| TNNC1 | Sarcomere, cardiac contraction | Missense | DCM, HCM |
| TNNT2 | Sarcomere, cardiac contraction | Missense | DCM, HCM, LVNC, RCM |
| TPM1 | Sarcomere, cardiac contraction | Truncating | DCM, HCM, LVNC |
| TTN | Sarcomere, cardiac contraction | Truncating | DCM, HCM, MD, MFM |
| VCL | Cytoskeleton | Truncating | DCM, HCM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spracklen, T.F.; Chakafana, G.; Schwartz, P.J.; Kotta, M.-C.; Shaboodien, G.; Ntusi, N.A.B.; Sliwa, K. Genetics of Peripartum Cardiomyopathy: Current Knowledge, Future Directions and Clinical Implications. Genes 2021, 12, 103. https://doi.org/10.3390/genes12010103
Spracklen TF, Chakafana G, Schwartz PJ, Kotta M-C, Shaboodien G, Ntusi NAB, Sliwa K. Genetics of Peripartum Cardiomyopathy: Current Knowledge, Future Directions and Clinical Implications. Genes. 2021; 12(1):103. https://doi.org/10.3390/genes12010103
Chicago/Turabian StyleSpracklen, Timothy F., Graham Chakafana, Peter J. Schwartz, Maria-Christina Kotta, Gasnat Shaboodien, Ntobeko A. B. Ntusi, and Karen Sliwa. 2021. "Genetics of Peripartum Cardiomyopathy: Current Knowledge, Future Directions and Clinical Implications" Genes 12, no. 1: 103. https://doi.org/10.3390/genes12010103
APA StyleSpracklen, T. F., Chakafana, G., Schwartz, P. J., Kotta, M.-C., Shaboodien, G., Ntusi, N. A. B., & Sliwa, K. (2021). Genetics of Peripartum Cardiomyopathy: Current Knowledge, Future Directions and Clinical Implications. Genes, 12(1), 103. https://doi.org/10.3390/genes12010103