Fetal Anomalies Associated with Novel Pathogenic Variants in TMEM94

 , , ,
, , ,  and
and 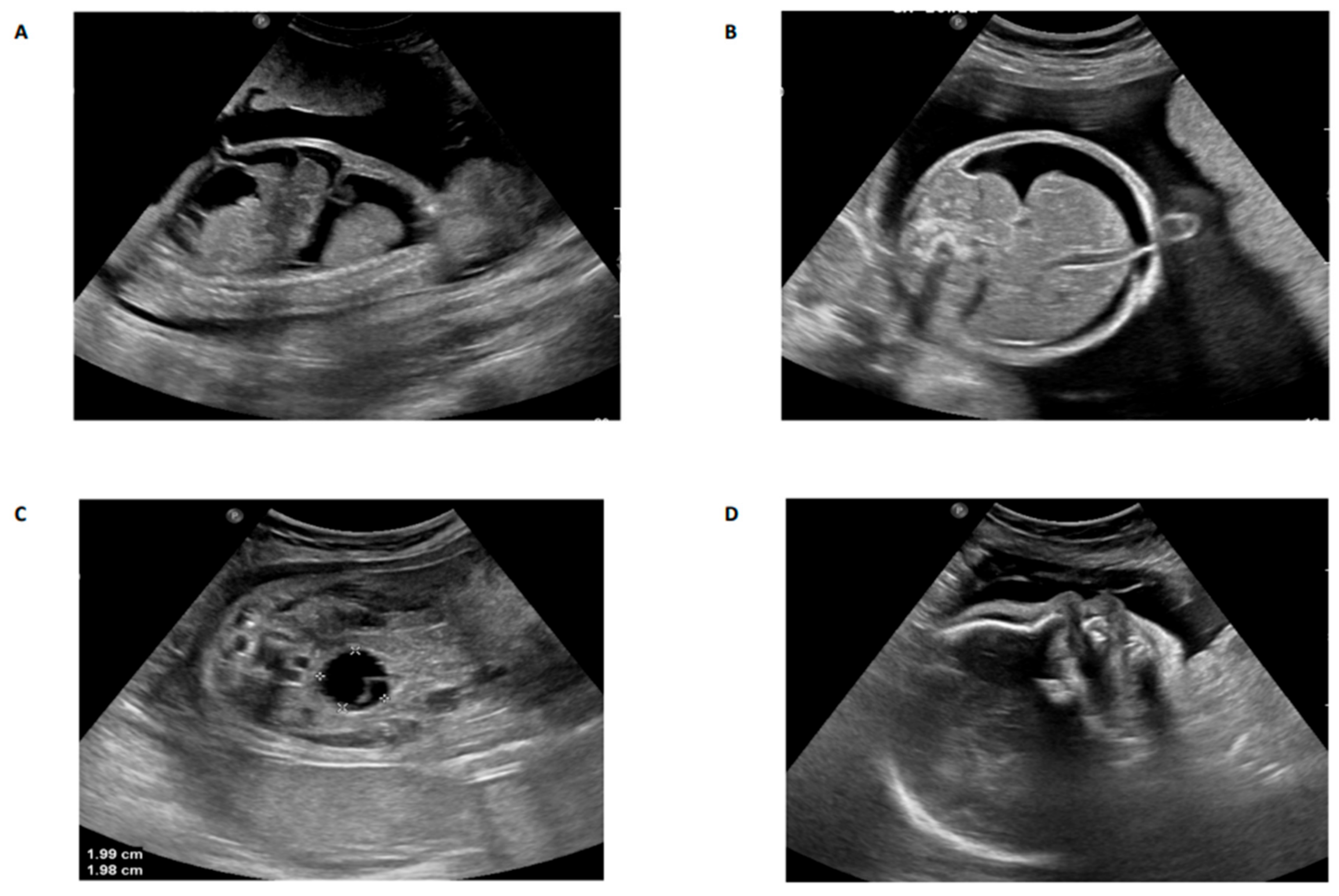{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
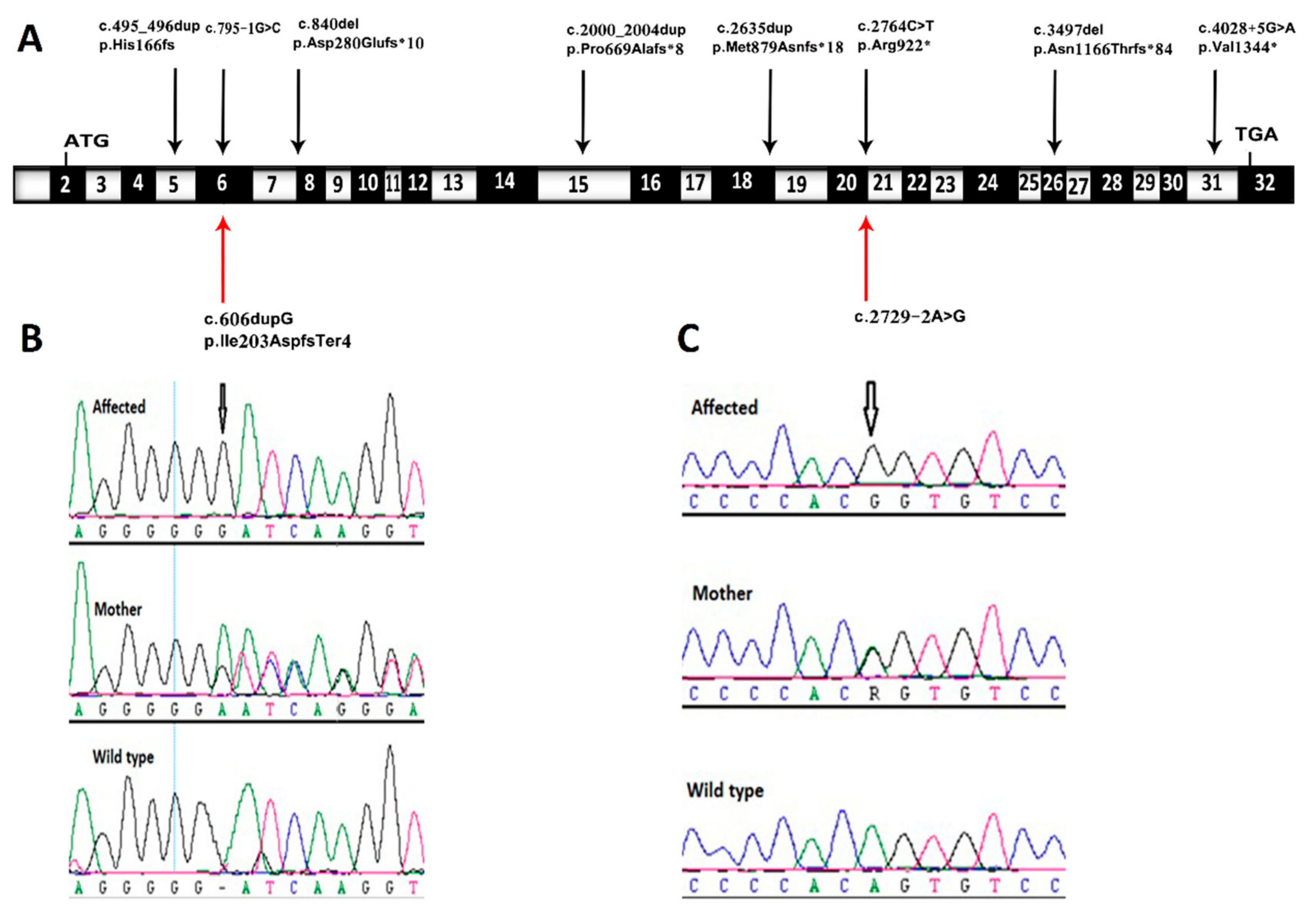
3. Results and Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stephen, J.; Maddirevula, S.; Nampoothiri, S.; Burke, J.D.; Herzog, M.; Shukla, A.; Steindl, K.; Eskin, A.; Patil, S.J.; Joset, P.; et al. Bi-allelic TMEM94 truncating variants are associated with neurodevelopmental delay, congenital heart defects, and distinct facial dysmorphism. Am. J. Hum. Genet. 2018, 103, 948–967. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Seki, N.; Ishikawa, K.; Tanaka, A.; Nomura, N. Prediction of the coding sequences of unidentified human genes. V. The coding sequences of 40 new genes (KIAA0161-KIAA0200) deduced by analysis of cDNA clones from human cell line KG-1 (supplement). DNA Res. 1996, 3, 43–53. [Google Scholar] [CrossRef][Green Version]
- Al-Hamed, M.H.; Kurdi, W.; Alsahan, N.; Alabdullah, Z.; Abudraz, R.; Tulbah, M.; Alnemer, M.; Khan, R.; Al-Jurayb, H.; Alahmed, A.; et al. Genetic spectrum of Saudi Arabian patients with antenatal cystic kidney disease and ciliopathy phenotypes using a targeted renal gene panel. J. Med. Genet. 2016, 53, 338–347. [Google Scholar] [CrossRef]
- Alkuraya, F.S. Genetics and genomic medicine in Saudi Arabia. Mol. Genet. Genom. Med. 2014, 2, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Abouelhoda, M.; AlSayed, M.; Alhassnan, Z.; Alotaibi, M.; Kayyali, H.; Al-Owain, M.; Shah, A.; Rahbeeni, Z.; Al-Muhaizea, M.A.; et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum. Genet. 2017, 136, 921–939. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Abouelhoda, M.; Assoum, M.; Moghrabi, N.; Rafiullah, R.; Almontashiri, N.; Alowain, M.; Alzaidan, H.; Alsayed, M.; Subhani, S.; et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am. J. Hum. Genet. 2019, 105, 879. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamed, M.H.; Alsahan, N.; Rice, S.J.; Edwards, N.; Nooreddeen, E.; Alotaibi, M.; Kurdi, W.; Alnemer, M.; Altaleb, N.; Ali, E.; et al. Bialleleic PKD1 mutations underlie early-onset autosomal dominant polycystic kidney disease in Saudi Arabian families. Pediatr. Nephrol. 2019, 34, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Ta-Shma, A.; Khan, T.N.; Vivante, A.; Willer, J.R.; Matak, P.; Jalas, C.; Pode-Shakked, B.; Salem, Y.; Anikster, Y.; Hildebrandt, F.; et al. Mutations in TMEM260 cause a pediatric neurodevelopmental, cardiac, and renal syndrome. Am. J. Hum. Genet. 2017, 100, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Silhavy, J.L.; Lee, J.E.; Al-Gazali, L.; Thomas, S.; Davis, E.E.; Bielas, S.L.; Hill, K.J.; Iannicelli, M.; Brancati, F.; et al. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science 2012, 335, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, F.; Amyere, M.; Jaeken, J.; Zeevaert, R.; Schollen, E.; Race, V.; Bammens, R.; Morelle, W.; Rosnoblet, C.; Legrand, D.; et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 2012, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Ansari, S.; Mardawi, E.A.; Alshammari, M.J.; Alkuraya, F.S. Mutations in TMEM231 cause Meckel-Gruber syndrome. J. Med. Genet. 2013, 50, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.X.; Ouwenga, R.; Anderson, R.L.; Waggoner, D.J.; Ober, C. A population-based study of autosomal-recessive disease-causing mutations in a founder population. Am. J. Hum. Genet. 2012, 91, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.M.; Consugar, M.; Tee, L.J.; McKee, B.M.; Maina, E.N.; Whelan, S.; Morgan, N.V.; Goranson, E.; Gissen, P.; Lilliquist, G.; et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat. Genet. 2006, 38, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Vuillaumier-Barrot, S.; Bouchet-Seraphin, C.; Chelbi, M.; Devisme, L.; Quentin, S.; Gazal, S.; Laquerrière, A.; Fallet-Bianco, C.; Loget, P.; Odent, S.; et al. Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am. J. Hum. Genet. 2012, 91, 1135–1143. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Hamed, M.H.; Alsahan, N.; Tulbah, M.; Kurdi, W.; Ali, W.I.; Sayer, J.A.; Imtiaz, F. Fetal Anomalies Associated with Novel Pathogenic Variants in TMEM94. Genes 2020, 11, 967. https://doi.org/10.3390/genes11090967
Al-Hamed MH, Alsahan N, Tulbah M, Kurdi W, Ali WI, Sayer JA, Imtiaz F. Fetal Anomalies Associated with Novel Pathogenic Variants in TMEM94. Genes. 2020; 11(9):967. https://doi.org/10.3390/genes11090967
Chicago/Turabian StyleAl-Hamed, Mohamed H., Nada Alsahan, Maha Tulbah, Wesam Kurdi, Wafa’a I. Ali, John A. Sayer, and Faiqa Imtiaz. 2020. "Fetal Anomalies Associated with Novel Pathogenic Variants in TMEM94" Genes 11, no. 9: 967. https://doi.org/10.3390/genes11090967
APA StyleAl-Hamed, M. H., Alsahan, N., Tulbah, M., Kurdi, W., Ali, W. I., Sayer, J. A., & Imtiaz, F. (2020). Fetal Anomalies Associated with Novel Pathogenic Variants in TMEM94. Genes, 11(9), 967. https://doi.org/10.3390/genes11090967