An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I

 , , and
, , and 
Abstract
1. Introduction
1.1. Myotonic Dystrophy Type I
1.2. Alternative Splicing
2. Muscleblind-Like (MBNL) Proteins and Mantaining MBNL/CELF1 Equilibrium in DM1
2.1. MBNL-Dependent Splicing Regulation
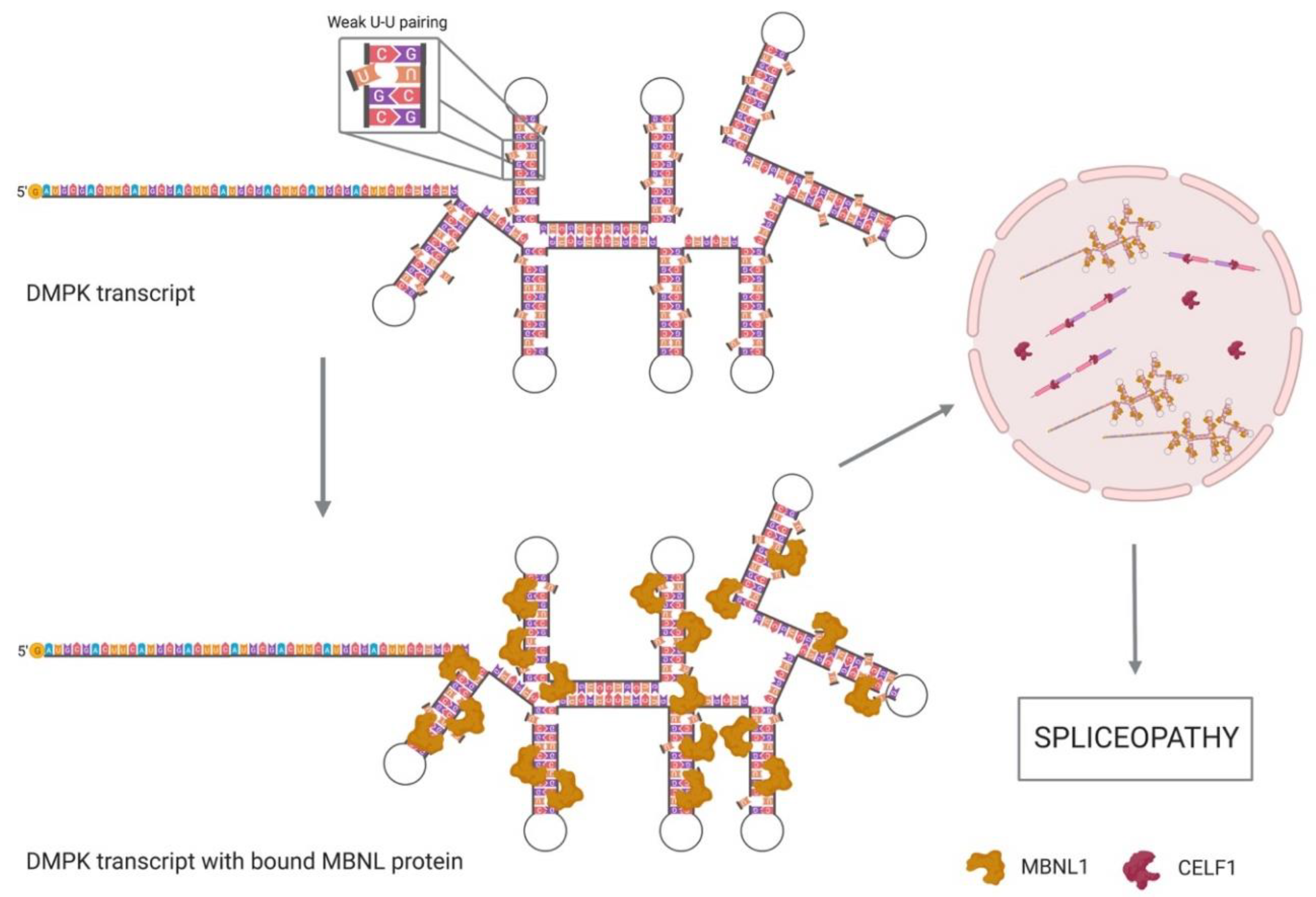
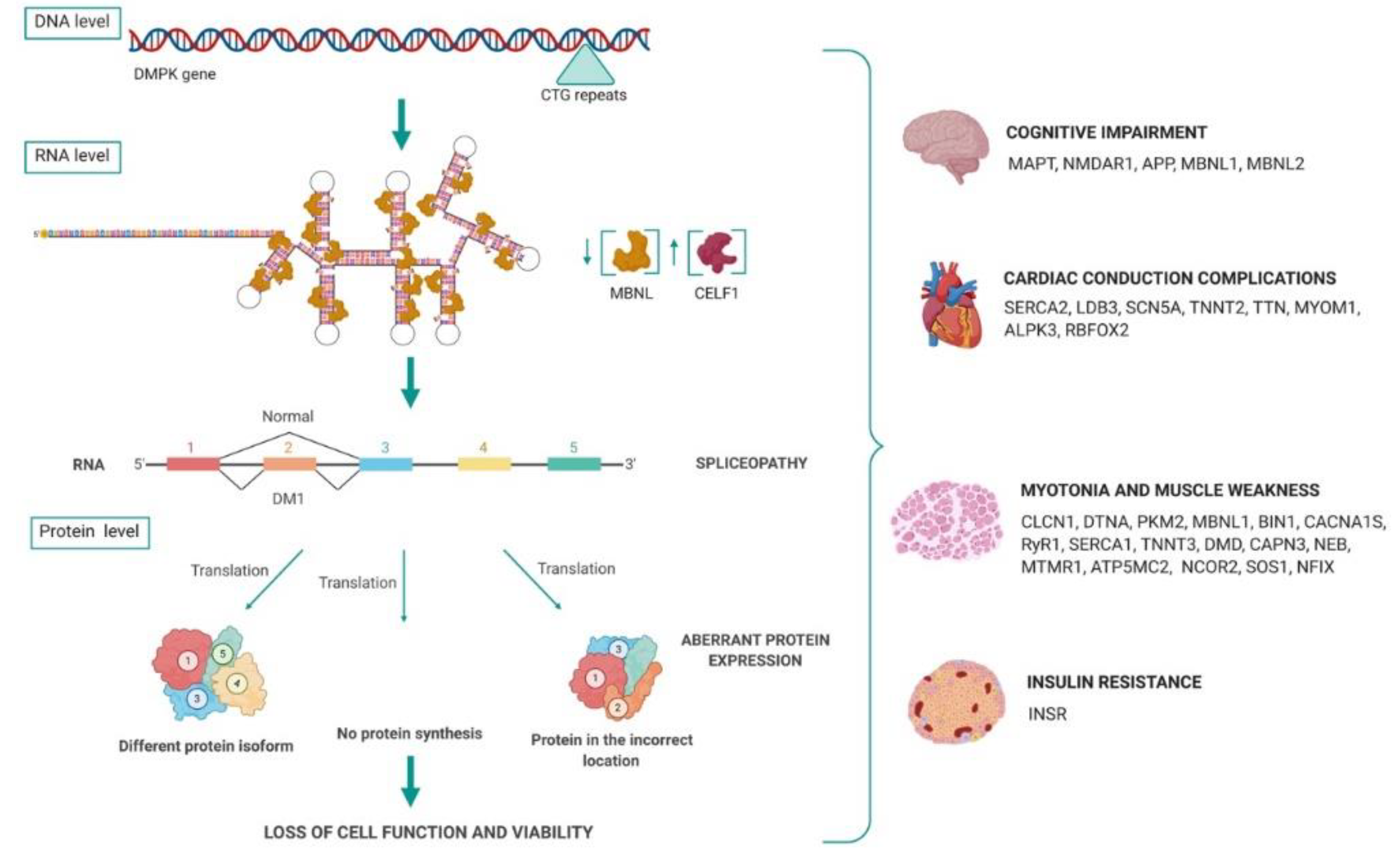
2.2. MBNL and CELF1 Implication in DM1
3. Spliceopathy Due to RNA Toxicity
3.1. Misregulation of mRNA Processing
3.1.1. Transcripts Altered in DM1 Brain Tissue
3.1.2. Transcripts Altered in DM1 Skeletal Muscle
3.1.3. Transcripts Altered in DM1 Cardiac Muscle
3.2. Misregulation of mRNA Localization and Stability
3.3. Misregulation of mRNA Translation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Day, J.W.; Ranum, L.P.W. Genetics and molecular pathogenesis of the myotonic dystrophies. Curr. Neurol. Neurosci. Rep. 2005, 5, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Yum, K.; Wang, E.T.; Kalsotra, A. Myotonic dystrophy: Disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr. Opin. Genet. Dev. 2017, 44, 30–37. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Wheeler, T.M. Myotonic Dystrophy: Therapeutic Strategies for the Future. Neurotherapeutics 2008. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef]
- Harper, P.S. The Genetic Basis of Myotonic Dystrophy. In Myotonic Dystrophy, 3rd ed.; London WB Saunders: London, UK, 2001; pp. 307–363. [Google Scholar]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 69, 385. [Google Scholar] [CrossRef]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Savic Pavicevic, D.; Miladinovic, J.; Brkusanin, M.; Svikovic, S.; Djurica, S.; Brajuskovic, G.; Romac, S. Molecular genetics and genetic testing in myotonic dystrophy type 1. Biomed. Res. Int. 2013, 2013, 391821. [Google Scholar] [CrossRef]
- Lavedan, C.; Hofmann-Radvanyi, H.; Shelbourne, P.; Rabes, J.P.; Duros, C.; Savoy, D.; Dehaupas, I.; Luce, S.; Johnson, K.; Junien, C. Myotonic dystrophy: Size- and sex-dependent dynamics of CTG meiotic instability, and somatic mosaicism. Am. J. Hum. Genet. 1993, 52, 875–883. [Google Scholar]
- Ballester-Lopez, A.; Linares-Pardo, I.; Koehorst, E.; Núñez-Manchón, J.; Pintos-Morell, G.; Coll-Cantí, J.; Almendrote, M.; Lucente, G.; Arbex, A.; Magaña, J.J.; et al. The need for establishing a universal CTG sizing method in myotonic dystrophy type 1. Genes 2020, 11, 757. [Google Scholar] [CrossRef]
- Botta, A.; Rossi, G.; Marcaurelio, M.; Fontana, L.; D’Apice, M.R.; Brancati, F.; Massa, R.; G Monckton, D.; Sangiuolo, F.; Novelli, G. Identification and characterization of 5′ CCG interruptions in complex DMPK expanded alleles. Eur. J. Hum. Genet. 2017, 25, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Braida, C.; Stefanatos, R.K.A.; Adam, B.; Mahajan, N.; Smeets, H.J.M.; Niel, F.; Goizet, C.; Arveiler, B.; Koenig, M.; Lagier-Tourenne, C.; et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2010. [Google Scholar] [CrossRef] [PubMed]
- Musova, Z.; Mazanec, R.; Krepelova, A.; Ehler, E.; Vales, J.; Jaklova, R.; Prochazka, T.; Koukal, P.; Marikova, T.; Kraus, J.; et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. Part A 2009. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Willems, P.; Coerwinkel, M.; Nillesen, W.; Smeets, H.; Vits, L.; Howeler, C.; Brunner, H.; Wieringa, B. Gonosomal mosaicism in myotonic dystrophy patients: Involvement of mitotic events in (CTG)(n) repeat variation and selection against extreme expansion in sperm. Am. J. Hum. Genet. 1994. [Google Scholar] [CrossRef]
- Wong, L.J.C.; Ashizawa, T.; Monckton, D.G.; Caskey, C.T.; Richards, C.S. Somatic heterogeneity of the CTG repeat in myotonic dystrophy is age and size dependent. Am. J. Hum. Genet. 1995, 56, 114–122. [Google Scholar]
- Thornton, C.A.; Johnson, K.; Moxley, R.T., III; Moxley, R.T. Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol. 1994, 35, 104–107. [Google Scholar] [CrossRef]
- Zatz, M.; Passos-bueno, M.R.; Cerqueira, A.; Marie, S.K.; Vainzof, M.; Pavanello, R.C.M. Analysis of the CTG repeat in skeletal muscle of young and adult myotonic dystrophy patients: When does the expansion occur? Hum. Mol. Genet. 1995. [Google Scholar] [CrossRef]
- Yin, Q.; Wang, H.; Li, N.; Ding, Y.; Xie, Z.; Jin, L.; Li, Y.; Wang, Q.; Liu, X.; Xu, L.; et al. Dosage effect of multiple genes accounts for multisystem disorder of myotonic dystrophy type 1. Cell Res. 2020, 30, 133–145. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). NCBI Homo Sapiens Annotation Release 108. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Homo_sapiens/108/#FeatureCountsStats (accessed on 2 April 2020).
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Tapial, J.; Ha, K.C.H.; Sterne-Weiler, T.; Gohr, A.; Braunschweig, U.; Hermoso-Pulido, A.; Quesnel-Vallières, M.; Permanyer, J.; Sodaei, R.; Marquez, Y.; et al. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef]
- Barash, Y.; Vaquero-Garcia, J.; González-Vallinas, J.; Xiong, H.Y.; Gao, W.; Lee, L.J.; Frey, B.J. AVISPA: A web tool for the prediction and analysis of alternative splicing. Genome Biol. 2013. [Google Scholar] [CrossRef]
- Fiszbein, A.; Kornblihtt, A.R. Alternative splicing switches: Important players in cell differentiation. BioEssays 2017, 39, 1600157. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008. [Google Scholar] [CrossRef] [PubMed]
- Laurent, F.-X.; Sureau, A.; Klein, A.F.; Trouslard, F.; Gasnier, E.; Furling, D.; Marie, J. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res. 2012, 40, 3159–3171. [Google Scholar] [CrossRef] [PubMed]
- Galka-Marciniak, P.; Urbanek, M.O.; Krzyzosiak, W.J. Triplet repeats in transcripts: Structural insights into RNA toxicity. Biol. Chem. 2012, 393, 1299–1315. [Google Scholar] [CrossRef]
- Nakamori, M.; Sobczak, K.; Puwanant, A.; Welle, S.; Eichinger, K.; Pandya, S.; Dekdebrun, J.; Heatwole, C.R.; McDermott, M.P.; Chen, T.; et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013, 74, 862–872. [Google Scholar] [CrossRef]
- Thomas, J.D.; Sznajder, L.J.; Bardhi, O.; Aslam, F.N.; Anastasiadis, Z.P.; Scotti, M.M.; Nishino, I.; Nakamori, M.; Wang, E.T.; Swanson, M.S. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017, 31, 1122–1133. [Google Scholar] [CrossRef]
- Wang, E.T.; Treacy, D.; Eichinger, K.; Struck, A.; Estabrook, J.; Olafson, H.; Wang, T.T.; Bhatt, K.; Westbrook, T.; Sedehizadeh, S.; et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lueck, J.D.; Lungu, C.; Mankodi, A.; Osborne, R.J.; Welle, S.L.; Dirksen, R.T.; Thornton, C.A. Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1. Am. J. Physiol. Cell Physiol. 2007, 292, 1291–1297. [Google Scholar] [CrossRef]
- Wang, E.T.; Cody, N.A.L.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide Regulation of Pre-mRNA Splicing and mRNA Localization by Muscleblind Proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Masuda, A.; Andersen, H.S.; Doktor, T.K.; Okamoto, T.; Ito, M.; Andresen, B.S.; Ohno, K. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci. Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Vicente, M.; Monferrer, L.; Artero, R. The Muscleblind family of proteins: An emerging class of regulators of developmentally programmed alternative splicing. Differentiation 2006, 74, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A muscleblind knockout model for myotonic dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef]
- Fardaei, M. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002, 11, 805–814. [Google Scholar] [CrossRef]
- Lee, K.S.; Cao, Y.; Witwicka, H.E.; Tom, S.; Tapscott, S.J.; Wang, E.H. RNA-binding protein muscleblind-like 3 (MBNL3) disrupts myocyte enhancer factor 2 (Mef2) β-exon splicing. J. Biol. Chem. 2010. [Google Scholar] [CrossRef]
- Dansithong, W.; Paul, S.; Comai, L.; Ready, S. Erratum: MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J. Biol. Chem. 2005, 280, 5773–5780. [Google Scholar] [CrossRef] [PubMed]
- Cass, D.; Hotchko, R.; Barber, P.; Jones, K.; Gates, D.P.; Berglund, J.A. The four Zn fingers of MBNL1 provide a flexible platform for recognition of its RNA binding elements. BMC Mol. Biol. 2011, 12, 20. [Google Scholar] [CrossRef]
- Tran, H.; Gourrier, N.; Lemercier-Neuillet, C.; Dhaenens, C.-M.; Vautrin, A.; Fernandez-Gomez, F.J.; Arandel, L.; Carpentier, C.; Obriot, H.; Eddarkaoui, S.; et al. Analysis of Exonic Regions Involved in Nuclear Localization, Splicing Activity, and Dimerization of Muscleblind-like-1 Isoforms. J. Biol. Chem. 2011, 286, 16435–16446. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Dong, S.; Choudhury, R.; Jin, Y.; Wang, Z. Treatment of type 1 myotonic dystrophy by engineering site-specific RNA endonucleases that target (CUG)(n) repeats. Mol. Ther. 2014, 22, 312–320. [Google Scholar] [CrossRef]
- Goers, E.S.; Purcell, J.; Voelker, R.B.; Gates, D.P.; Berglund, J.A. MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res. 2010, 38, 2467–2484. [Google Scholar] [CrossRef] [PubMed]
- Charizanis, K.; Lee, K.Y.; Batra, R.; Goodwin, M.; Zhang, C.; Yuan, Y.; Shiue, L.; Cline, M.; Scotti, M.M.; Xia, G.; et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 2012, 75, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Costa, J.M.; Llamusi, M.B.; Garcia-Lopez, A.; Artero, R. Alternative splicing regulation by Muscleblind proteins: From development to disease. Biol. Rev. 2011, 86, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, F.; Ladd, A.N. Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biol. 2010, 7, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Compton, S.A.; Sobczak, K.; Stenberg, M.G.; Thornton, C.A.; Griffith, J.D.; Swanson, M.S. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007, 35, 5474–5486. [Google Scholar] [CrossRef]
- Kiliszek, A.; Kierzek, R.; Krzyzosiak, W.J.; Rypniewski, W. Atomic resolution structure of CAG RNA repeats: Structural insights and implications for the trinucleotide repeat expansion diseases. Nucleic Acids Res. 2010. [Google Scholar] [CrossRef]
- Taylor, K.; Sznajder, Ł.J.; Cywoniuk, P.; Thomas, J.D.; Swanson, M.S.; Sobczak, K. MBNL splicing activity depends on RNA binding site structural context. Nucleic Acids Res. 2018, 46, 9119–9133. [Google Scholar] [CrossRef]
- Du, H.; Cline, M.S.; Osborne, R.J.; Tuttle, D.L.; Clark, T.A.; Donohue, J.P.; Hall, M.P.; Shiue, L.; Swanson, M.S.; Thornton, C.A.; et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010, 17, 187–193. [Google Scholar] [CrossRef]
- Osborne, R.J.; Lin, X.; Welle, S.; Sobczak, K.; O’Rourke, J.R.; Swanson, M.S.; Thornton, C.A. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 2009, 18, 1471–1481. [Google Scholar] [CrossRef]
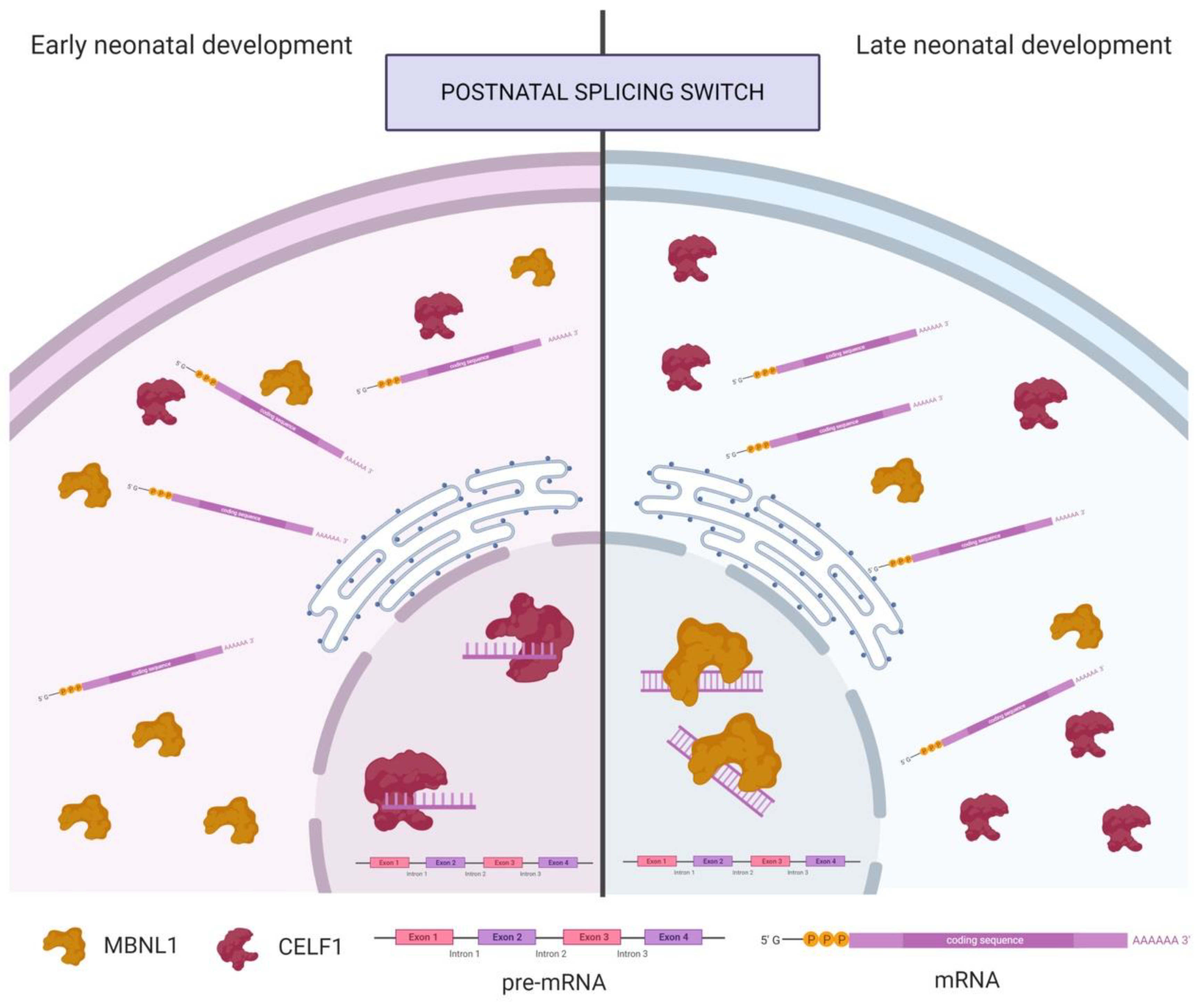
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.H.; Charlet-B, N.; Poulos, M.G.; Singh, G.; Swanson, M.S.; Cooper, T.A. Muscleblind proteins regulate alternative splicing. EMBO J. 2004, 23, 3103–3112. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Stepniak-Konieczna, E.; Sobczak, K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014, 42, 10873–10887. [Google Scholar] [CrossRef]
- Bargiela, A.; Llamusi, B.; Cerro-Herreros, E.; Artero, R. Two enhancers control transcription of Drosophila muscleblind in the embryonic somatic musculature and in the central nervous system. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Sasagawa, N.; Suzuki, K.; Ishiura, S. The CUG-binding protein binds specifically to UG dinucleotide repeats in a yeast three-hybrid system. Biochem. Biophys. Res. Commun. 2000. [Google Scholar] [CrossRef]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef]
- Kino, Y. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum. Mol. Genet. 2004, 13, 495–507. [Google Scholar] [CrossRef][Green Version]
- Van Cruchten, R.T.P.; Wieringa, B.; Wansink, D.G. Expanded CUG repeats in DMPK transcripts adopt diverse hairpin conformations without influencing the structure of the flanking sequences. RNA 2019, 25, 481–495. [Google Scholar] [CrossRef]
- Jahromi, A.H.; Fu, Y.; Miller, K.A.; Nguyen, L.; Luu, L.M.; Baranger, A.M.; Zimmerman, S.C. Developing bivalent ligands to target CUG triplet repeats, the causative agent of myotonic dystrophy type 1. J. Med. Chem. 2013, 56, 9471–9481. [Google Scholar] [CrossRef]
- Sobczak, K.; de Mezer, M.; Michlewski, G.; Krol, J.; Krzyzosiak, W.J. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003. [Google Scholar] [CrossRef]
- Sznajder, Ł.J.; Michalak, M.; Taylor, K.; Cywoniuk, P.; Kabza, M.; Wojtkowiak-Szlachcic, A.; Matłoka, M.; Konieczny, P.; Sobczak, K. Mechanistic determinants of MBNL activity. Nucleic Acids Res. 2016, 44, 10326–10342. [Google Scholar] [CrossRef] [PubMed]
- Querido, E.; Gallardo, F.; Beaudoin, M.; Ménard, C.; Chartrand, P. Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J. Cell Sci. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska, M.; Taylor, K.; Sobczak, K.; Napierala, M.; Krzyzosiak, W.J. Small molecule kinase inhibitors alleviate different molecular features of myotonic dystrophy type 1. RNA Biol. 2014, 11, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Tapscott, S.J. Myotonic dystrophy: Emerging mechanisms for DM1 and DM2. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, L. Correction of RNA-binding protein CUGBP1 and gsk3β signaling as therapeutic approach for congenital and adult myotonic dystrophy type 1. Int. J. Mol. Sci. 2020, 21, 94. [Google Scholar] [CrossRef]
- Timchenko, N.A.; Iakova, P.; Cai, Z.J.; Smith, J.R.; Timchenko, L.T. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol. Cell. Biol. 2001, 21, 6927–6938. [Google Scholar] [CrossRef]
- Jones, K.; Jin, B.; Iakova, P.; Huichalaf, C.; Sarkar, P.; Schneider-Gold, C.; Schoser, B.; Meola, G.; Shyu, A.-B.; Timchenko, N.; et al. RNA Foci, CUGBP1, and ZNF9 Are the Primary Targets of the Mutant CUG and CCUG Repeats Expanded in Myotonic Dystrophies Type 1 and Type 2. Am. J. Pathol. 2011, 179, 2475–2489. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef]
- Misra, C.; Lin, F.; Kalsotra, A. Deregulation of RNA metabolism in microsatellite expansion diseases. Adv. Neurobiol. 2018, 20, 213–238. [Google Scholar] [CrossRef]
- Davis, B.M.; McCurrach, M.E.; Taneja, K.L.; Singer, R.H.; Housman, D.E. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. USA 1997, 94, 7388–7393. [Google Scholar] [CrossRef] [PubMed]
- Malatesta, M.; Giagnacovo, M.; Cardani, R.; Meola, G.; Pellicciari, C. RNA processing is altered in skeletal muscle nuclei of patients affected by myotonic dystrophy. Histochem. Cell Biol. 2011, 135, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Huichalaf, C.; Sakai, K.; Jin, B.; Jones, K.; Wang, G.L.; Schoser, B.; Schneider-Gold, C.; Sarkar, P.; Pereira-Smith, O.M.; Timchenko, N.; et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J. 2010. [Google Scholar] [CrossRef] [PubMed]
- Orengo, J.P.; Ward, A.J.; Cooper, T.A. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann. Neurol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Thornell, L.E.; Lindstöm, M.; Renault, V.; Klein, A.; Mouly, V.; Ansved, T.; Butler-Browne, G.; Furling, D. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol. Appl. Neurobiol. 2009. [Google Scholar] [CrossRef]
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef]
- Bachinski, L.L.; Baggerly, K.A.; Neubauer, V.L.; Nixon, T.J.; Raheem, O.; Sirito, M.; Unruh, A.K.; Zhang, J.; Nagarajan, L.; Timchenko, L.T.; et al. Most expression and splicing changes in myotonic dystrophy type 1 and type 2 skeletal muscle are shared with other muscular dystrophies. Neuromuscul. Disord. 2014, 24, 227–240. [Google Scholar] [CrossRef]
- Furling, D. Misregulation of alternative splicing and microRNA processing in DM1 pathogenesis. Rinsho Shinkeigaku 2012, 52, 1018–1022. [Google Scholar] [CrossRef][Green Version]
- Chau, A.; Kalsotra, A. Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics. Dev. Dyn. 2015, 377–390. [Google Scholar] [CrossRef]
- Ladd, A.N. CUG-BP, Elav-like family (CELF)-mediated alternative splicing regulation in the brain during health and disease. Mol. Cell. Neurosci. 2013, 56, 456–464. [Google Scholar] [CrossRef]
- Goodwin, M.; Mohan, A.; Batra, R.; Lee, K.Y.; Charizanis, K.; Fernández Gómez, F.J.; Eddarkaoui, S.; Sergeant, N.; Buée, L.; Kimura, T.; et al. MBNL Sequestration by Toxic RNAs and RNA Misprocessing in the Myotonic Dystrophy Brain. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gomez, F.; Tran, H.; Dhaenens, C.-M.; Caillet-Boudin, M.-L.; Schraen-Maschke, S.; Blum, D.; Sablonnière, B.; Buée-Scherrer, V.; Buee, L.; Sergeant, N. Myotonic Dystrophy: An RNA Toxic Gain of Function Tauopathy? In Tau Biology; Springer: Singapore, 2019; pp. 207–216. [Google Scholar]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Matsuura, T.; Shinmi, J.; Amakusa, Y.; Masuda, A.; Ito, M.; Kinoshita, M.; Furuya, H.; Abe, K.; Ibi, T.; et al. Four parameters increase the sensitivity and specificity of the exon array analysis and disclose 25 novel aberrantly spliced exons in myotonic dystrophy. J. Hum. Genet. 2012, 57, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Dhaenens, C.M.; Schraen-Maschke, S.; Tran, H.; Vingtdeux, V.; Ghanem, D.; Leroy, O.; Delplanque, J.; Vanbrussel, E.; Delacourte, A.; Vermersch, P.; et al. Overexpression of MBNL1 fetal isoforms and modified splicing of Tau in the DM1 brain: Two individual consequences of CUG trinucleotide repeats. Exp. Neurol. 2008, 210, 467–478. [Google Scholar] [CrossRef]
- Blake, D.J.; Nawrotzki, R.; Peters, M.F.; Froehner, S.C.; Davies, K.E. Isoform diversity of dystrobrevin, the murine 87-kDa postsynaptic protein. J. Biol. Chem. 1996. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Cooper, T.A. Reexpression of pyruvate kinase M2 in type 1 myofibers correlates with altered glucose metabolism in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 13570–13575. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). DTNA dystrobrevin α [Homo sapiens (human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/1837 (accessed on 16 April 2020).
- Anselmo, H.-I.J.; Samuel, L.-C.M.; Ricardo, M.-G.; Natalie, S.-G.; Bulmaro, C. Localization of α-Dystrobrevin in Cajal Bodies and Nucleoli: A New Role for α-Dystrobrevin in the Structure/Stability of the Nucleolus. J. Cell. Biochem. 2015, 116, 2755–2765. [Google Scholar] [CrossRef]
- Peters, M.F.; Sadoulet-Puccio, H.M.; Grady, R.M.; Kramarcy, N.R.; Kunkel, L.M.; Sanes, J.R.; Sealock, R.; Froehner, S.C. Differential membrane localization and intermolecular associations of α-dystrobrevin isoforms in skeletal muscle. J. Cell Biol. 1998, 142, 1269–1278. [Google Scholar] [CrossRef]
- Charlet, B.N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG Repeats Trigger Aberrant Splicing of ClC-1 Chloride Channel Pre-mRNA and Hyperexcitability of Skeletal Muscle in Myotonic Dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Chen, T.T.; Klassen, T.L.; Goldman, A.M.; Marini, C.; Guerrini, R.; Noebels, J.L. Novel brain expression of ClC-1 chloride channels and enrichment of CLCN1 variants in epilepsy. Neurology 2013, 80, 1078–1085. [Google Scholar] [CrossRef]
- Fugier, C.; Klein, A.F.; Hammer, C.; Vassilopoulos, S.; Ivarsson, Y.; Toussaint, A.; Tosch, V.; Vignaud, A.; Ferry, A.; Messaddeq, N.; et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011, 17, 720–725. [Google Scholar] [CrossRef]
- Tang, Z.Z.; Yarotskyy, V.; Wei, L.; Sobczak, K.; Nakamori, M.; Eichinger, K.; Moxley, R.T.; Dirksen, R.T.; Thornton, C.A. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum. Mol. Genet. 2012, 21, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Nakamori, M.; Lueck, J.D.; Pouliquin, P.; Aoike, F.; Fujimura, H.; Dirksen, R.T.; Takahashi, M.P.; Dulhunty, A.F.; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005, 14, 2189–2200. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Urbinati, C.R.; Crusselle, V.J.; Luo, D.; Lee, Y.J.; Harrison, J.K.; Oh, S.P.; Swanson, M.S. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr. Patterns 2003. [Google Scholar] [CrossRef]
- Traverso, M.; Assereto, S.; Baratto, S.; Iacomino, M.; Pedemonte, M.; Diana, M.C.; Ferretti, M.; Broda, P.; Minetti, C.; Gazzerro, E.; et al. Clinical and molecular consequences of exon 78 deletion in DMD gene. J. Hum. Genet. 2018, 63, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Koebis, M.; Ohsawa, N.; Kino, Y.; Sasagawa, N.; Nishino, I.; Ishiura, S. Alternative splicing of myomesin 1 gene is aberrantly regulated in myotonic dystrophy type 1. Genes Cells 2011, 16, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Ottenheijm, C.A.C.; Witt, C.C.; Stienen, G.J.; Labeit, S.; Beggs, A.H.; Granzier, H. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum. Mol. Genet. 2009. [Google Scholar] [CrossRef]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef]
- Buj-Bello, A. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum. Mol. Genet. 2002, 11, 2297–2307. [Google Scholar] [CrossRef]
- Chen, G.; Masuda, A.; Konishi, H.; Ohkawara, B.; Ito, M.; Kinoshita, M.; Kiyama, H.; Matsuura, T.; Ohno, K. Phenylbutazone induces expression of MBNL1 and suppresses formation of MBNL1-CUG RNA foci in a mouse model of myotonic dystrophy. Sci. Rep. 2016, 6, 25317. [Google Scholar] [CrossRef] [PubMed]
- Lueck, J.D.; Mankodi, A.; Swanson, M.S.; Thornton, C.A.; Dirksen, R.T. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J. Gen. Physiol. 2007, 129, 79–94. [Google Scholar] [CrossRef]
- Rau, F.; Laine, J.; Ramanoudjame, L.; Ferry, A.; Arandel, L.; Delalande, O.; Jollet, A.; Dingli, F.; Lee, K.Y.; Peccate, C.; et al. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat. Commun. 2015, 6, 7205. [Google Scholar] [CrossRef] [PubMed]
- Groh, W.J.; Groh, M.R.; Saha, C.; Kincaid, J.C.; Simmons, Z.; Ciafaloni, E.; Pourmand, R.; Otten, R.F.; Bhakta, D.; Nair, G.V.; et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N. Engl. J. Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, A.; Varin, J.; Babuty, D.; rédéric Anselme, F.; Coste, J.; Duboc, D. Long-term follow-up of arrhythmias in patients with myotonic dystrophy treated by pacing: A multicenter diagnostic pacemaker study. J. Am. Coll. Cardiol. 2002. [Google Scholar] [CrossRef]
- Freyermuth, F.; Rau, F.; Kokunai, Y.; Linke, T.; Sellier, C.; Nakamori, M.; Kino, Y.; Arandel, L.; Jollet, A.; Thibault, C.; et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun. 2016, 7, 11067. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.M.; Choi, J.; El-Ghazali, A.; Park, S.Y.; Roos, K.P.; Jordan, M.C.; Fishbein, M.C.; Comai, L.; Reddy, S. Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci. Rep. 2015, 5, 9042. [Google Scholar] [CrossRef]
- Yamashita, Y.; Matsuura, T.; Kurosaki, T.; Amakusa, Y.; Kinoshita, M.; Ibi, T.; Sahashi, K.; Ohno, K. LDB3 splicing abnormalities are specific to skeletal muscles of patients with myotonic dystrophy type 1 and alter its PKC binding affinity. Neurobiol. Dis. 2014, 69, 200–205. [Google Scholar] [CrossRef]
- Misra, C.; Bangru, S.; Lin, F.; Lam, K.; Koenig, S.N.; Lubbers, E.R.; Hedhli, J.; Murphy, N.P.; Parker, D.J.; Dobrucki, L.W.; et al. Aberrant Expression of a Non-muscle RBFOX2 Isoform Triggers Cardiac Conduction Defects in Myotonic Dystrophy. Dev. Cell 2020, 52, 748–763.e6. [Google Scholar] [CrossRef]
- Taliaferro, J.M.; Vidaki, M.; Oliveira, R.; Olson, S.; Zhan, L.; Saxena, T.; Wang, E.T.; Graveley, B.R.; Gertler, F.B.; Swanson, M.S.; et al. Distal Alternative Last Exons Localize mRNAs to Neural Projections. Mol. Cell 2016, 61, 821–833. [Google Scholar] [CrossRef]
- Wang, P.-Y.Y.; Chang, K.-T.T.; Lin, Y.-M.M.; Kuo, T.-Y.Y.; Wang, G.-S.S. Ubiquitination of MBNL1 Is Required for Its Cytoplasmic Localization and Function in Promoting Neurite Outgrowth. Cell Rep. 2018, 22, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, S. Increased nuclear but not cytoplasmic activities of CELF1 protein leads to muscle wasting. Hum. Mol. Genet. 2020, 1–15. [Google Scholar] [CrossRef]
- Rattenbacher, B.; Beisang, D.; Wiesner, D.L.; Jeschke, J.C.; von Hohenberg, M.; St. Louis-Vlasova, I.A.; Bohjanen, P.R. Analysis of CUGBP1 Targets Identifies GU-Repeat Sequences That Mediate Rapid mRNA Decay. Mol. Cell. Biol. 2010, 30, 3970–3980. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Lee, J.E.; López, C.M.; Anderson, J.; Nguyen, T.M.P.; Heck, A.M.; Wilusz, J.; Wilusz, C.J. The CELF1 RNA-Binding protein regulates decay of signal recognition particle mRNAs and limits secretion in mouse myoblasts. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.; Mittal, S.; Furling, D.; Butler-Browne, G.S.; David Brook, J.; Morris, G.E. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells 2007, 12, 1035–1048. [Google Scholar] [CrossRef]
- St.Louis-Vlasova, I.; Bohjanen, P.R. Coordinate regulation of mRNA decay networks by GU-rich elements and CELF1. Curr. Opin. Genet. Dev. 2011, 21, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Ambrose, K.K.; Ishak, T.; Lian, L.-H.; Goh, K.-J.; Wong, K.-T.; Ahmad-Annuar, A.; Thong, M.-K. Deregulation of microRNAs in blood and skeletal muscles of myotonic dystrophy type 1 patients. Neurol. India 2017, 65, 512–517. [Google Scholar] [CrossRef]
- Fritegotto, C.; Ferrati, C.; Pegoraro, V.; Angelini, C. Micro-RNA expression in muscle and fiber morphometry in myotonic dystrophy type 1. Neurol. Sci. 2017. [Google Scholar] [CrossRef]
- Perfetti, A.; Greco, S.; Bugiardini, E.; Cardani, R.; Gaia, P.; Gaetano, C.; Meola, G.; Martelli, F. Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromuscul. Disord. 2014, 24, 509–515. [Google Scholar] [CrossRef][Green Version]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.D.; Ranum, L.P.W. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum. Mol. Genet. 2013, 22, R45–R51. [Google Scholar] [CrossRef] [PubMed]
- Mondragon-Gonzalez, R.; Perlingeiro, R.C.R. Recapitulating muscle disease phenotypes with myotonic dystrophy 1 iPS cells: A tool for disease modeling and drug discovery. Dis. Model Mech. 2018. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Splicing Alteration | Reference | Tissue Expression | Implications in DM1 Pathology | Sample Type | |
|---|---|---|---|---|---|---|
| Exon/Intron | Inclusion/Exclusion | |||||
| APP | Exon 7 | Exclusion | Jiang (2004) [85] | Brain | n.d. | DM1 patients brain sections |
| MAPT | Exon 2 | Exclusion | Goodwin (2015) [83] | Brain (frontal cortex) | Progressive appearance of NFTs composed of intraneuronal aggregates of hyperphosphorylated tau protein. | DM1 patients brain sections |
| Exon 3 | Exclusion | |||||
| Exon 10 | Exclusion | |||||
| MBNL1 | Exon 6 | Inclusion | Dhaenens (2008) [87] | Most tissues | Splicing defects | DM1 patients brain sections |
| Exon 8 | Inclusion | |||||
| MBNL2 | Exon 7 | Inclusion | Nakamori (2013) [29] | Brain | Splicing defects | DM1 patient-derived cultured myotubes |
| Exon 8 | Inclusion | Yamashita (2012) [86] | ||||
| NMDAR1 | Exon 5 | Inclusion | Jiang (2004) [85] | Brain | Memory impairment. | DM1 patients brain sections |
| Gene | Splicing Alteration | Reference | Tissue Expression | Implications in DM1 Pathology | Sample Type | |
|---|---|---|---|---|---|---|
| Exon/Intron | Inclusion/Exclusion | |||||
| ALPK3 | Exon 2 | Inclusion | Nakamori (2013) [29] | Cardiac muscle | Not described | DM1 patient skeletal muscle biopsy |
| ATP2A1 | Exon 22 | Exclusion | Kimura (2005) [98] | Skeletal muscle | Muscle degeneration: impair intracellular calcium homeostasis | DM1 patient-derived cultured myotubes |
| ATP5MC2 | Exon 1 | Inclusion | Yamashita (2012) [86] | Skeletal muscle | n.d. | DM1 patient skeletal muscle biopsy |
| BIN1 | Exon 11 | Exclusion | Fugier (2011) [96] | Skeletal muscle | Muscle weakness: altered excitation–contraction coupling | DM1 patient skeletal muscle biopsy |
| CACNA1S | Exon 29 | Exclusion | Tang (2012) [97] | Skeletal muscle | Muscle weakness: altered excitation–contraction coupling | DM1 patient skeletal muscle biopsy |
| CAPN3 | Exon 16 | Exclusion | Yamashita (2012) [86] | Skeletal muscle | Muscle weakness: decreased protease activity | DM1 patient skeletal muscle biopsy |
| CLCN1 | Intron 2 | Inclusion | Charlet (2002) [93] Mankodi (2002) [94] | Skeletal muscle Brain | Myotonia | DM1 patient skeletal muscle biopsy |
| Exon 7a | Inclusion | Lueck (2006) [106] Nakamori (2013) [29] | ||||
| DMD | Exon 71 | Exclusion | Yamashita (2012) [86] | Skeletal muscle | Muscle weakness: alteration of the membrane integrity | DM1 patient-derived cultured myotubes |
| Exon 78 | Exclusion | Rau (2015) [107] | ||||
| DTNA | Exon 11a | Exclusion | Nakamori (2013) [29] | Brain Cardiac muscle Skeletal muscle | Muscle weakness | DM1 patient skeletal muscle biopsy |
| Exon 12 | Exclusion | |||||
| FHOD | Exon 11a | Exclusion | Yamashita (2012) [86] | Skeletal muscle | n.d. | DM1 patient skeletal muscle biopsy |
| GFPT1 | Exon 9 | Exclusion | Nakamori (2013) [29] | Most tissues | n.d. | DM1 patient skeletal muscle biopsy |
| INSR | Exon 11 | Exclusion | Savkur (2001) [103] | Skeletal muscle | Insulin resistance: decreased metabolic response to insulin | DM1 patient skeletal muscle biopsy and cultured myotubes derived from DM1 patient fibroblasts |
| MBNL1 | Exon 5 | Inclusion | Konieczny (2014) [55] | Skeletal muscle | Splicing defects | DM1 patient skeletal muscle biopsy |
| Exon 7 | Inclusion | Nakamori (2013) [29] | ||||
| Exon 10 | Inclusion | Yamashita (2012) [86] | ||||
| MYOM1 | Exon 17a | Inclusion | Koebis (2011) [101] | Skeletal muscle | Sarcomeric M-band instability | DM1 patient skeletal muscle biopsy |
| MTMR1 | Exon 2.1 | Inclusion | Buj-Bello (2002) [104] | Skeletal muscle | Impaired myogenesis | DM1 patient-derived cultured myotubes |
| Exon 2.2 | Inclusion | Yamashita (2012) [86] | ||||
| Exon 2.3 | Inclusion | Buj-Bello (2002) [104] | ||||
| MXRA7 | Exon 4 | Exclusion | Yamashita (2012) [86] | Most tissues | n.d. | DM1 patient skeletal muscle biopsy |
| NCOR2 | Exon 10 | Inclusion | Yamashita (2012) [86] | Skeletal muscle | n.d. | DM1 patient skeletal muscle biopsy |
| NEB | Exon 116 | Inclusion | Yamashita (2012) [86] | Skeletal muscle | n.d. | DM1 patient skeletal muscle biopsy |
| NFIX | Exon 7 | Inclusion | Yamashita (2012) [86] | Skeletal muscle | n.d. | DM1 patient skeletal muscle biopsy |
| NRAP | Exon 12 | Exclusion | Lin (2006) [52] | Skeletal muscle | Altered myofibril assembly. | DM1 patient skeletal muscle biopsy |
| PKM2 | Exon 9 | Inclusion | Gao (2013) [89] | Brain Type I fibres from skeletal muscle in DM1 | Defects in energy metabolism in skeletal muscle | DM1 patient skeletal muscle biopsy |
| Exon 10 | Inclusion | |||||
| RYR1 | Exon 70 | Exclusion | Kimura (2005) [98] | Skeletal muscle | Muscle weakness: decreased muscle contraction | DM1 patient-derived cultured myotubes |
| SMYD1 | Exon 39 | Inclusion | Du (2010) [50] | Skeletal muscle Cardiac muscle | n.d. | DM1 patient skeletal muscle biopsy |
| SOS1 | Exon 25 | Exclusion | Yamashita (2012) [86] | Skeletal muscle | Inhibits signalling pathways involved in muscle hypertrophy | DM1 patient skeletal muscle biopsy |
| TNNT3 | Exon 23 | Inclusion | Yamashita (2012) [86] | Skeletal muscle | Alteration of the sarcomere structure | DM1 patient skeletal muscle biopsy |
| Gene | Splicing Alteration | Reference | Tissue Expression | Implications in DM1 Pathology | Sample Type | |
|---|---|---|---|---|---|---|
| Exon/Intron | Inclusion/Exclusion | |||||
| ATP2A2 | Intron 19 | Inclusion | Kimura (2005) [98] Dixon (2015) [111] | Cardiac muscle | Cardiac conduction impairment: deregulated calcium influx | DM1 patient-derived cultured myotubes |
| RBFOX2 | 3 nt | Exclusion | Misra (2020) [113] | Cardiac muscle | Cardiac conduction delay and arrhythmogenesis | DM1 patient cardiac muscle tissue |
| SCN5A | Exon 6a | Inclusion | Freyermuth (2016) [110] | Cardiac muscle | Conduction slowing: decreased upstroke of the cardiac action potential | DM1 patient cardiac muscle tissue |
| TNNT2 | Exon 5 | Inclusion | Ho (2004) [54] Dixon (2015) [111] | Cardiac muscle | Alteration of the contractile properties: different calcium sensitivity of the myofilament | DM1 patient skeletal muscle biopsy |
| TTN | Zr4 | Inclusion | Lin (2006) [52] Yamashita (2012) [86] | Cardiac muscle | Defective myofibril assembly and function | DM1 patient skeletal muscle biopsy |
| Zr5 | Inclusion | |||||
| Mex5 | Inclusion | |||||
| ZASP/LDB | Exon 5 | Inclusion | Nakamori (2013) [29] | Cardiac muscle | Morphological abnormalities of the cardiac fibre | DM1 patient skeletal muscle biopsy |
| Exon 11 | Inclusion | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Martínez, A.; Soblechero-Martín, P.; de-la-Puente-Ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes 2020, 11, 1109. https://doi.org/10.3390/genes11091109
López-Martínez A, Soblechero-Martín P, de-la-Puente-Ovejero L, Nogales-Gadea G, Arechavala-Gomeza V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes. 2020; 11(9):1109. https://doi.org/10.3390/genes11091109
Chicago/Turabian StyleLópez-Martínez, Andrea, Patricia Soblechero-Martín, Laura de-la-Puente-Ovejero, Gisela Nogales-Gadea, and Virginia Arechavala-Gomeza. 2020. "An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I" Genes 11, no. 9: 1109. https://doi.org/10.3390/genes11091109
APA StyleLópez-Martínez, A., Soblechero-Martín, P., de-la-Puente-Ovejero, L., Nogales-Gadea, G., & Arechavala-Gomeza, V. (2020). An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes, 11(9), 1109. https://doi.org/10.3390/genes11091109