Mapping QTL Associated with Resistance to Avian Oncogenic Marek’s Disease Virus (MDV) Reveals Major Candidate Genes and Variants




Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.1.1. Full Sib Advanced Inter-Cross Line (FSIL)
2.1.2. Pure line Pedigreed Sire Populations with Daughter Progeny Records for MDV Mortality
2.2. Genomic Sequencing of Founder Birds of the FSIL
2.3. Annotation of Genome Variants
2.4. 600 K High Density SNP Genotyping of F6 Birds
2.5. GWAS of the F6 Population to Identify MD Resistance QTL
2.6. Identification of QTL and Their Confidence Intervals
2.7. RNA-Seq Analysis
2.7.1. MDV Challenge Experiment
2.7.2. RNA Preparation
2.7.3. Transcriptomic Sequencing and Analysis
2.8. QTLR Tests
2.8.1. Selection of Candidate Genomic Elements Underlying the 38 QTLR for Further Testing and Validation in the 8 HL Elite Pure Lines
2.8.2. Identification of Candidate Genes within QTLR
2.8.3. Selection of Genetic Markers within QTLR for Genotyping
2.8.4. Selection of Candidate Coding Genes
2.8.5. Selection of Candidate Non-Coding RNAs
2.8.6. Selection of Potentially Functional Variants in Coding Genes
2.8.7. Selecting QTLR Markers from the Candidate Genomic Elements, for Genotyping in the 8 Elite HL Pure Lines
2.9. Genotyping of the Eight Elite Lines
2.10. QTLR Association Analysis
3. Results
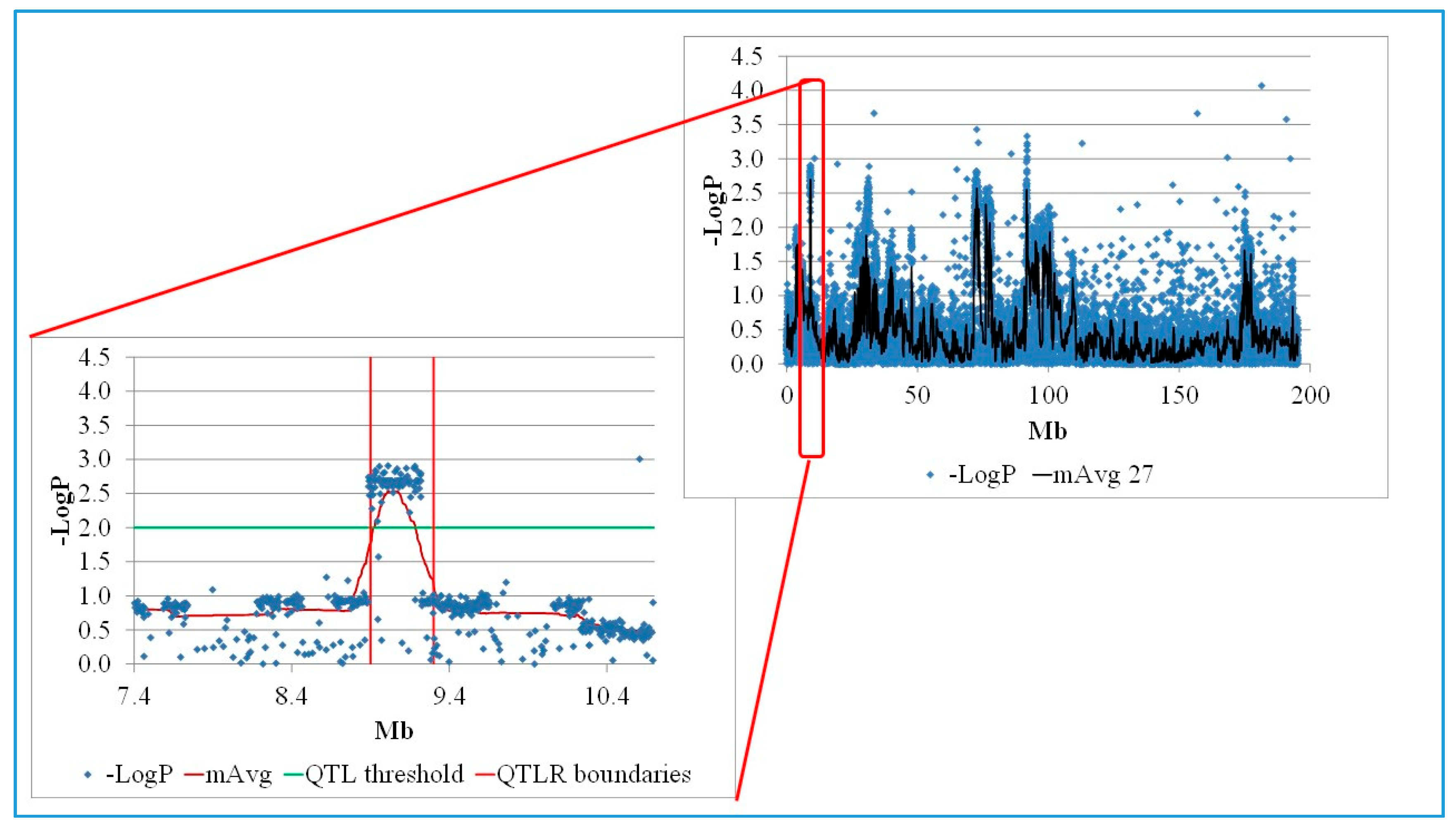
3.1. High Resolution MD QTL Mapping in Advanced Full-Sib Inter-Cross F6 Families
3.2. Annotation of QTLR
3.3. Identification and Analysis of SNPs in F0 Parental Birds
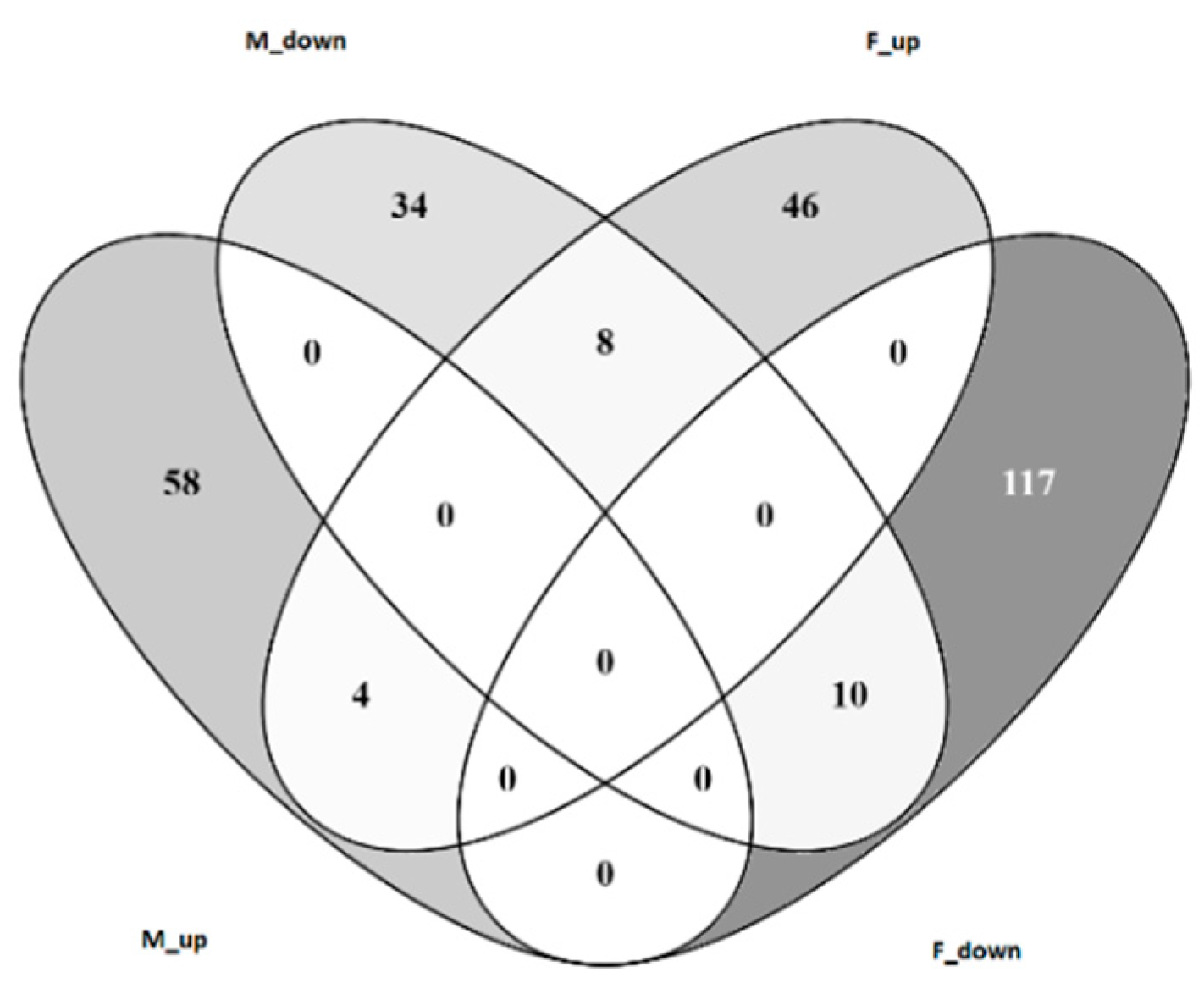
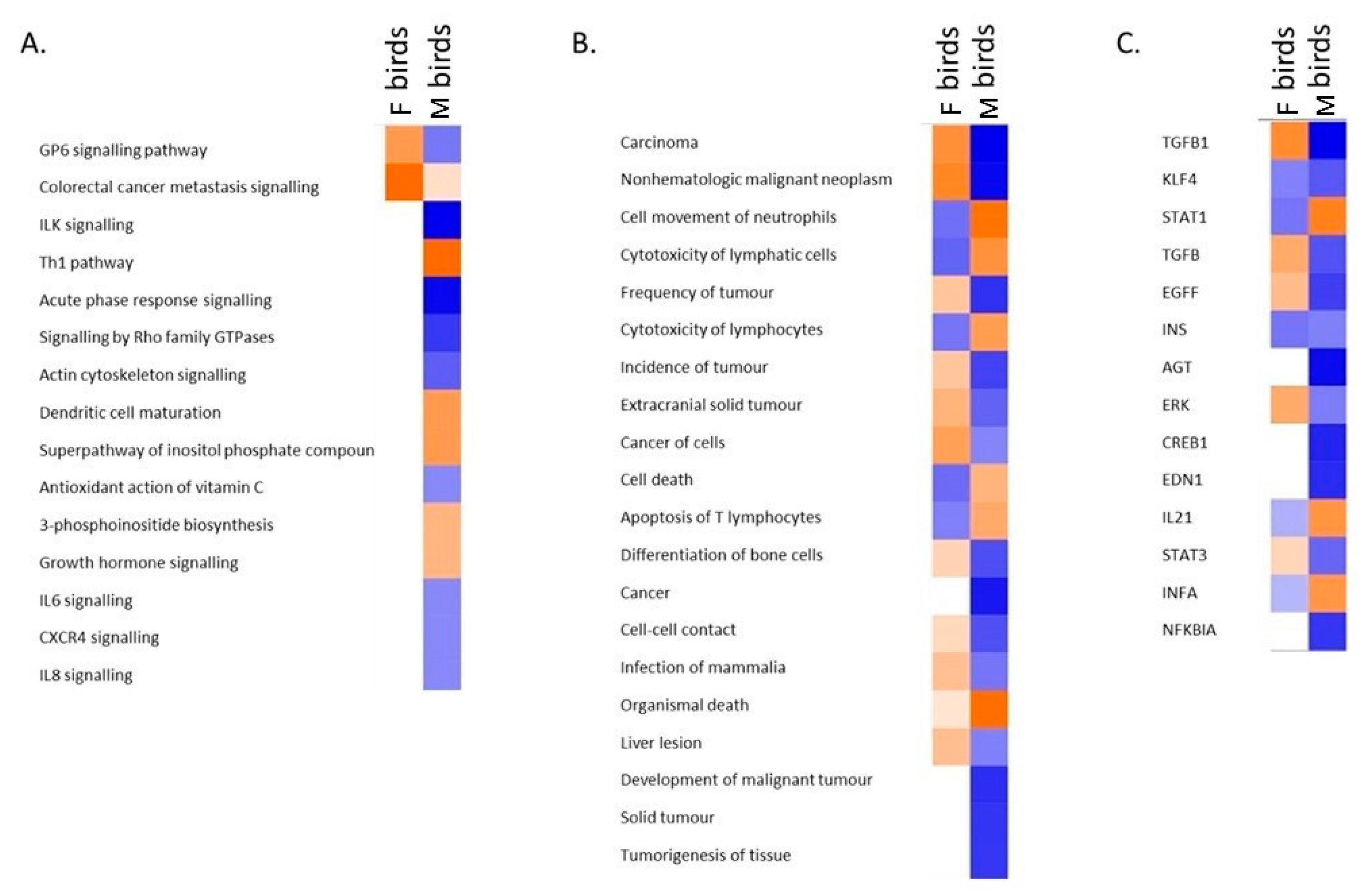
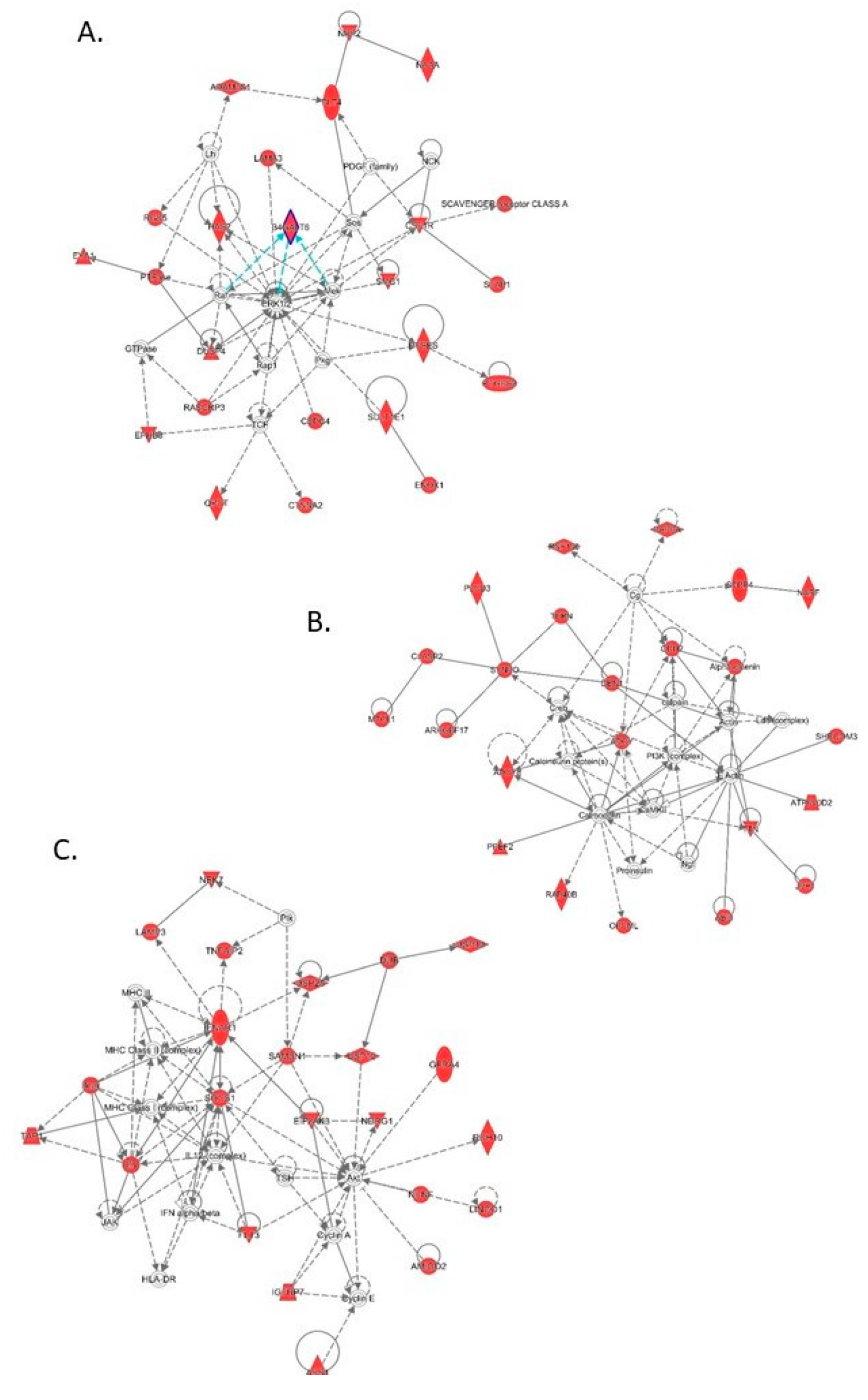
3.4. Transcriptomic Analysis of Male and Female Commercial Birds Challenged with MDV
3.5. Analysis of Differentially Expressed Genes Located in QTLR
3.6. Validating QTLR for Association with MDV Resistance
3.6.1. Populations, Genomic Elements and Markers
3.6.2. Genetic Association Tests
3.6.3. Summary of Element x Line Tests
3.7. Validation of QTLR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Approval
Availability of Data
References
- Morrow, C.; Fehler, F. Marek’s disease: A worldwide problem. In Marek’s Disease, An Evolving Problem; Davison, F., Nair, V., Eds.; Elsevier Ltd.: Oxford, UK, 2004; pp. 49–61. [Google Scholar]
- Nair, V. Evolution of Marek’s disease—A paradigm for incessant race between the pathogen and the host. Vet. J. 2005, 170, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.F.; Wong, C.W.; Walkden-Brown, S.W.; Colditz, I.G.; Arzey, K.E.; Groves, P.J. Immunosuppressive effects of Marek’s disease virus (MDV) and herpesvirus of turkeys (HVT) in broiler chickens and the protective effect of HVT vaccination against MDV challenge. Avian Pathol. 2002, 31, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Witter, R.L. Protective efficacy of Marek’s disease vaccines. Curr. Top. Microbiol. Immunol. 2001, 255, 57–90. [Google Scholar] [CrossRef] [PubMed]
- Read, A.F.; Baigent, S.J.; Powers, C.; Kgosana, L.B.; Blackwell, L.; Smith, L.P.; Kennedy, D.A.; Walkden-Brown, S.W.; Nair, V.K. Imperfect Vaccination Can Enhance the Transmission of Highly Virulent Pathogens. PLoS Biol. 2015, 13, e1002198. [Google Scholar] [CrossRef] [PubMed]
- Boodhoo, N.; Gurung, A.; Sharif, S.; Behboudi, S. Marek’s disease in chickens: A review with focus on immunology. Vet. Res. 2016, 47, 119. [Google Scholar] [CrossRef]
- Fulton, J.E.; Arango, J.; Arthur, J.A.; Settar, P.; Kreager, K.S.; O’Sullivan, N.P. Improving the outcome of a Marek’s disease challenge in multiple lines of egg type chickens. Avian Dis. 2013, 57 (Suppl. S2), 519–522. [Google Scholar] [CrossRef]
- Wolc, A.; Arango, J.; Jankowski, T.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Fernando, R.; Garrick, D.J.; Dekkers, J.C. Genome-wide association study for Marek’s disease mortality in layer chickens. Avian Dis. 2013, 57 (Suppl. S2), 395–400. [Google Scholar] [CrossRef]
- Burgess, S.C.; Young, J.R.; Baaten, B.J.; Hunt, L.; Ross, L.N.; Parcells, M.S.; Kumar, P.M.; Tregaskes, C.A.; Lee, L.F.; Davison, T.F. Marek’s disease is a natural model for lymphomas overexpressing Hodgkin’s disease antigen (CD30). Proc. Natl. Acad. Sci. USA 2004, 101, 13879–13884. [Google Scholar] [CrossRef]
- Li, K.; Lian, L.; Yang, N.; Qu, L. Temporal expression and DNA hypomethylation profile of CD30 in Marek’s disease virus-infected chicken spleens. Poult. Sci. 2015, 94, 1165–1169. [Google Scholar] [CrossRef]
- Bacon, L.D.; Witter, R.L.; Crittenden, L.B.; Fadly, A.; Motta, J. B-haplotype influence on Marek’s disease, Rous sarcoma, and lymphoid leukosis virus-induced tumors in chickens. Poult. Sci. 1981, 60, 1132–1139. [Google Scholar] [CrossRef]
- Kaufman, J. The simple chicken major histocompatibility complex: Life and death in the face of pathogens and vaccines. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Camp, S.; Collen, T.; Avila, D.; Salomonsen, J.; Wallny, H.-J.; van Hateren, A.; Hunt, L.; Jacob, J.P.; Johnston, F.; et al. Structures of an MHC class I molecule from B21 chickens illustrate promiscuous peptide binding. Immunity 2007, 27, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Hunt, H.D.; Jadhao, S.; Swayne, D.E. Major histocompatibility complex and background genes in chickens influence susceptibility to high pathogenicity avian influenza virus. Avian Dis. 2010, 54 (Suppl. S1), 572–575. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Kung, H.J.; Fulton, J.E.; Morgan, R.W.; Cheng, H.H. Growth hormone interacts with the Marek’s disease virus SORF2 protein and is associated with disease resistance in chicken. Proc. Natl. Acad. Sci. USA 2001, 98, 9203–9208. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Cheng, H.H.; Tirunagaru, V.; Sofer, L.; Burnside, J. A strategy to identify positional candidate genes conferring Marek’s disease resistance by integrating DNA microarrays and genetic mapping. Anim. Genet. 2001, 32, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Niikura, M.; Fulton, J.E.; Cheng, H.H. Identification of chicken lymphocyte antigen 6 complex, locus E (LY6E, alias SCA2) as a putative Marek’s disease resistance gene via a virus-host protein interaction screen. Cytogenet. Genome Res. 2003, 102, 304–308. [Google Scholar] [CrossRef]
- Smith, J.; Sadeyen, J.R.; Paton, I.R.; Hocking, P.M.; Salmon, N.; Fife, M.; Nair, V.; Burt, D.W.; Kaiser, P. Systems analysis of immune responses in Marek’s disease virus-infected chickens identifies a gene involved in susceptibility and highlights a possible novel pathogenicity mechanism. J. Virol. 2011, 85, 11146–11158. [Google Scholar] [CrossRef]
- Meydan, H.; Yildiz, M.A.; Dodgson, J.B.; Cheng, H.H. Allele-specific expression analysis reveals CD79B has a cis-acting regulatory element that responds to Marek’s disease virus infection in chickens. Poult. Sci. 2011, 90, 1206–1211. [Google Scholar] [CrossRef]
- Li, D.F.; Lian, L.; Qu, L.J.; Chen, Y.M.; Liu, W.B.; Chen, S.R.; Zheng, J.X.; Xu, G.Y.; Yang, N. A genome-wide SNP scan reveals two loci associated with the chicken resistance to Marek’s disease. Anim. Genet. 2013, 44, 217–222. [Google Scholar] [CrossRef]
- Wang, D.; Sun, S.; Heidari, M. Marek’s disease vaccine activates chicken macrophages. J. Vet. Sci. 2018, 19, 375–383. [Google Scholar] [CrossRef]
- Chakraborty, P.; Kuo, R.; Vervelde, L.; Dutia, B.M.; Kaiser, P.; Smith, J. Macrophages from Susceptible and Resistant Chicken Lines have Different Transcriptomes following Marek’s Disease Virus Infection. Genes 2019, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; He, Y.; Zhang, L.; Ding, Y.; Lian, L.; Zhao, C.; Song, J.; Yang, N. Long intergenic non-coding RNA GALMD3 in chicken Marek’s disease. Sci. Rep. 2017, 7, 10294. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Zhang, Q.; Liu, C.; Song, J.; Yang, N.; Lian, L. Integrated analysis of lncRNA and mRNA repertoires in Marek’s disease infected spleens identifies genes relevant to resistance. BMC Genom. 2019, 20, 245. [Google Scholar] [CrossRef]
- Tian, F.; Luo, J.; Zhang, H.; Chang, S.; Song, J. MiRNA expression signatures induced by Marek’s disease virus infection in chickens. Genomics 2012, 99, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Parnas, O.; Corcoran, D.L.; Cullen, B.R. Analysis of the mRNA targetome of microRNAs expressed by Marek’s disease virus. MBio 2014, 5, e01060-13. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.A.; Liu, H.C. Impact of HVT Vaccination on Splenic miRNA Expression in Marek’s Disease Virus Infections. Genes 2019, 10, 115. [Google Scholar] [CrossRef]
- Neerukonda, S.N.; Tavlarides-Hontz, P.; McCarthy, F.; Pendarvis, K.; Parcells, M.S. Comparison of the Transcriptomes and Proteomes of Serum Exosomes from Marek’s Disease Virus-Vaccinated and Protected and Lymphoma-Bearing Chickens. Genes 2019, 10, 116. [Google Scholar] [CrossRef]
- Luo, J.; Mitra, A.; Tian, F.; Chan, S.; Zhang, H.; Cui, K.; Yu, Y.; Zhao, K.; Song, J. Histone methylation analysis and pathway predictions in chickens after MDV infection. PLoS ONE 2012, 7, e41849. [Google Scholar] [CrossRef]
- Mitra, A.; Luo, J.; He, Y.; Gu, Y.; Zhang, H.; Zhao, K.; Cui, K.; Song, J. Histone modifications induced by MDV infection at early cytolytic and latency phases. BMC Genom. 2015, 16, 311. [Google Scholar] [CrossRef]
- Heifetz, E.M.; Fulton, J.E.; O’Sullivan, N.P.; Arthur, J.; Cheng, H.; Wang, J.; Soller, M.; Dekkers, J.C.M. Mapping QTL affecting resistance to Marek’s disease in an F6 advanced intercross population of commercial layer chickens. BMC Genom. 2009, 10, 20. [Google Scholar] [CrossRef]
- Kranis, A.; Gheyas, A.A.; Boschiero, C.; Turner, F.; Yu, L.; Smith, S.; Talbot, R.; Pirani, A.; Brew, F.; Kaiser, P.; et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genom. 2013, 14, 59. [Google Scholar] [CrossRef]
- McElroy, J.P.; Dekkers, J.C.; Fulton, J.E.; O’Sullivan, N.P.; Soller, M.; Lipkin, E.; Zhang, W.; Koehler, K.J.; Lamont, S.J.; Cheng, H.H. Microsatellite markers associated with resistance to Marek’s disease in commercial layer chickens. Poult. Sci. 2005, 84, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- Heifetz, E.M.; Fulton, J.E.; O’Sullivan, N.P.; Arthur, J.A.; Wang, J.; Dekkers, J.C.; Soller, M. Mapping quantitative trait loci affecting susceptibility to Marek’s disease virus in a backcross population of layer chickens. Genetics 2007, 177, 2417–2431. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 October 2013).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; le Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Mosig, M.O.; Lipkin, E.; Khutoreskaya, G.; Tchourzyna, E.; Soller, M.; Friedmann, A. A whole genome scan for quantitative trait loci affecting milk protein percentage in Israeli-Holstein cattle, by means of selective milk DNA pooling in a daughter design, using an adjusted false discovery rate criterion. Genetics 2001, 157, 1683–1698. [Google Scholar]
- Fernando, R.L.; Nettleton, D.; Southey, B.R.; Dekkers, J.C.; Rothschild, M.F.; Soller, M. Controlling the proportion of false positives in multiple dependent tests. Genetics 2004, 166, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Lipkin, E.; Strillacci, M.G.; Eitam, H.; Yishay, M.; Schiavini, F.; Soller, M.; Bagnato, A.; Shabtay, A. The Use of Kosher Phenotyping for Mapping QTL Affecting Susceptibility to Bovine Respiratory Disease. PLoS ONE 2016, 11, e0153423. [Google Scholar] [CrossRef]
- Weller, J.I. Quantitative Trait Loci Analysis in Animals, 2nd ed.; CABI Publishing: London, UK, 2009. [Google Scholar]
- Kuo, R.I.; Tseng, E.; Eory, L.; Paton, I.R.; Archibald, A.L.; Burt, D.W. Normalized long read RNA sequencing in chicken reveals transcriptome complexity similar to human. BMC Genom. 2017, 18, 323. [Google Scholar] [CrossRef]
- Biggs, P.M.; Payne, L.N. Studies on Marek’s Disease. I. Experimental Transmission. JNCI J. Natl. Cancer Inst. 1967, 39, 267–280. [Google Scholar] [CrossRef]
- Khare, M.L.; Grun, J.; Adams, E.V. Marek’s disease in Japanese quail—A pathological, virological and serological study. Poult. Sci. 1975, 54, 2066–2068. [Google Scholar] [CrossRef] [PubMed]
- Bettridge, J.M.; Lynch, S.E.; Brena, M.C.; Melese, K.; Dessie, T.; Terfa, Z.G.; Desta, T.T.; Rushton, S.; Hanotte, O.; Kaiser, P.; et al. Infection-interactions in Ethiopian village chickens. Prev. Vet. Med. 2014, 117, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Perumbakkam, S.; Muir, W.M.; Black-Pyrkosz, A.; Okimoto, R.; Cheng, H.H. Comparison and contrast of genes and biological pathways responding to Marek’s disease virus infection using allele-specific expression and differential expression in broiler and layer chickens. BMC Genom. 2013, 14, 64. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef]
- Semagn, K.; Beyene, Y.; Warburton, M.L.; Tarekegne, A.; Mugo, S.; Meisel, B.; Sehabiague, P.; Prasanna, B.M. Meta-analyses of QTL for grain yield and anthesis silking interval in 18 maize populations evaluated under water-stressed and well-watered environments. BMC Genom. 2013, 14, 313. [Google Scholar] [CrossRef]
- Fulton, J.E.; McCarron, A.M.; Lund, A.R.; Pinegar, K.N.; Wolc, A.; Chazara, O.; Bed’Hom, B.; Berres, M.; Miller, M.M. A high-density SNP panel reveals extensive diversity, frequent recombination and multiple recombination hotspots within the chicken major histocompatibility complex B region between BG2 and CD1A1. Genet. Sel. Evol. 2016, 48, 1. [Google Scholar] [CrossRef]
- Vallejo, R.L.; Bacon, L.D.; Liu, H.C.; Witter, R.L.; Groenen, M.A.; Hillel, J.; Cheng, H.H. Genetic mapping of quantitative trait loci affecting susceptibility to Marek’s disease virus induced tumors in F2 intercross chickens. Genetics 1998, 148, 349–360. [Google Scholar] [PubMed]
- Yonash, N.; Bacon, L.D.; Witter, R.L.; Cheng, H.H. High resolution mapping and identification of new quantitative trait loci (QTL) affecting susceptibility to Marek’s disease. Anim. Genet. 1999, 30, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Niikura, M.; Kim, T.; Mao, W.; MacLea, K.S.; Hunt, H.; Dodgson, J.; Burnside, J.; Morgan, R.; Ouyang, M.; et al. Using integrative genomics to elucidate genetic resistance to Marek’s disease in chickens. Dev. Biol. 2008, 132, 365–372. [Google Scholar] [CrossRef]
- Goto, R.M.; Wang, Y.; Taylor, R.L., Jr.; Wakenell, P.S.; Hosomichi, K.; Shiina, T.; Blackmore, C.S.; Briles, W.E.; Miller, M.M. BG1 has a major role in MHC-linked resistance to malignant lymphoma in the chicken. Proc. Natl. Acad. Sci. USA 2009, 106, 16740–16745. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Tian, J.; Yang, X.; Pan, W.; Xu, B.; Li, B.; Ingvarsson, P.K.; Zhang, D. Identification of additive, dominant, and epistatic variation conferred by key genes in cellulose biosynthesis pathway in Populus tomentosa. DNA Res. 2015, 22, 53–67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pei, G.; Sun, H.; Dai, Y.; Liu, X.; Zhao, Z. Investigation of multi-trait associations using pathway-based analysis of GWAS summary statistics. BMC Genom. 2019, 20, 79. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTLR | Chr | Start | End | Length | Genes | lncRNAs |
|---|---|---|---|---|---|---|
| 1 | 1 | 8,854,589 | 9,261,317 | 406,729 | 0 | 1 |
| 2 | 1 | 13,268,810 | 14,294,877 | 1,026,068 | 12 | 34 |
| 3 | 1 | 52,012,381 | 52,517,953 | 505,573 | 2 | 4 |
| 4 | 1 | 71,738,977 | 73,085,956 | 1,346,980 | 16 | 28 |
| 5 | 1 | 75,288,912 | 77,765,499 | 2,476,588 | 86 | 79 |
| 6 | 1 | 91,482,501 | 91,803,475 | 320,975 | 10 | 9 |
| 7 | 1 | 101,761,945 | 104,477,713 | 2,715,769 | 31 | 37 |
| 8 | 1 | 109,395,871 | 110,913,451 | 1,517,581 | 20 | 32 |
| 9 | 1 | 169,656,958 | 172,228,091 | 2,571,134 | 32 | 44 |
| 10 | 1 | 174,394,951 | 175,634,386 | 1,239,436 | 23 | 33 |
| 11 | 1 | 194,193,118 | 194,788,548 | 595,431 | 18 | 23 |
| 12 | 2 | 15,960 | 883,257 | 867,298 | 45 | 20 |
| 13 | 2 | 46,423,161 | 46,868,789 | 445,629 | 11 | 12 |
| 14 | 2 | 105,493,833 | 108,988,920 | 3,495,088 | 34 | 47 |
| 15 | 2 | 125,168,753 | 127,089,304 | 1,920,552 | 25 | 34 |
| 16 | 2 | 138,767,830 | 139,701,774 | 933,945 | 4 | 7 |
| 17 | 3 | 108,220,206 | 109,260,649 | 1,040,444 | 13 | 9 |
| 18 | 4 | 8,308,498 | 11,268,107 | 2,959,610 | 54 | 99 |
| 19 | 4 | 84,393,173 | 88,579,400 | 4,186,228 | 68 | 73 |
| 20 | 5 | 7,568,851 | 8,147,837 | 578,987 | 4 | 3 |
| 21 | 5 | 18,806,924 | 19,673,354 | 866,431 | 4 | 3 |
| 22 | 6 | 2,077,640 | 2,709,412 | 631,773 | 19 | 13 |
| 23 | 6 | 29,536,109 | 29,817,337 | 281,229 | 10 | 16 |
| 24 | 6 | 31,006,769 | 31,448,342 | 441,574 | 7 | 6 |
| 25 | 7 | 13,062,871 | 16,436,053 | 3,373,183 | 55 | 82 |
| 26 | 10 | 22,643 | 1,713,384 | 1,690,742 | 63 | 28 |
| 27 | 11 | 7,397,790 | 8,440,259 | 1,042,470 | 9 | 28 |
| 28 | 12 | 8,996,686 | 9,432,693 | 436,008 | 15 | 16 |
| 29 | 13 | 10,363,430 | 12,176,727 | 1,813,298 | 27 | 40 |
| 30 | 14 | 8,085,563 | 9,335,685 | 1,250,123 | 38 | 50 |
| 31 | 14 | 13,138,194 | 15,087,518 | 1,949,325 | 82 | 41 |
| 32 | 16 | 1630 | 490,907 | 489,278 | 47 | 14 |
| 33 | 17 | 3,442,598 | 5,634,042 | 2,191,445 | 64 | 84 |
| 34 | 18 | 3,196,488 | 4,093,129 | 896,642 | 22 | 32 |
| 35 | 24 | 4,489,675 | 5,514,833 | 1,025,159 | 42 | 32 |
| 36 | 26 | 4,378,168 | 5,036,699 | 658,532 | 22 | 17 |
| 37 | 27 | 1,540,112 | 2,270,461 | 730,350 | 23 | 11 |
| 38 | 28 | 1,282,726 | 1,571,011 | 288,286 | 15 | 12 |
| QTLR | Gene | Description | Chr | Start | End | Length |
|---|---|---|---|---|---|---|
| 5 | CSTA | Cystatin A (Stefin A) | 1 | 76,253,583 | 76,258,549 | 4967 |
| 5 | LAG3 | Lymphocyte-activation gene 3 | 1 | 76,676,057 | 76,678,087 | 2031 |
| 5 | C1S | Complement C1s subcomponent | 1 | 76,859,359 | 76,868,459 | 9101 |
| 7 | ADAMTS5 | ADAM metallopeptidase with thrombospondin type 1 motif, 5 | 1 | 102,403,453 | 102,442,746 | 39,294 |
| 8 | SUPT20H | Spt20 homolog, saga complex component | 1 | 171,574,831 | 171,612,194 | 37,364 |
| 10 | FLT3 | Fms related tyrosine kinase 3 | 1 | 175,281,894 | 175,309,466 | 27,573 |
| 11 | RELT | RELT tumor necrosis factor receptor | 1 | 194,551,973 | 194,555,607 | 3635 |
| 13 | TRANK1 | Tetratricopeptide repeat and ankyrin repeat containing 1 | 2 | 46,537,531 | 46,576,714 | 39,184 |
| 19 | CTNNA2 | Gallus gallus catenin (cadherin-associated protein), α 2 | 4 | 86,948,459 | 87,057,215 | 108,757 |
| 29 | HAVCR1 | Hepatitis A virus cellular receptor 1 precursor | 13 | 10,778,915 | 10,785,137 | 6223 |
| 29 | TIMD4 | T-cell immunoglobulin and mucin domain-containing protein 4 | 13 | 10,789,088 | 10,803,683 | 14,596 |
| 30 | SOCS1 | Suppressor of cytokine signaling 1 | 14 | 8,935,374 | 8,936,082 | 709 |
| 32 | TAP1 | Antigen peptide transporter 1 | 16 | 67,623 | 76,124 | 8502 |
| 32 | IL4I1 | Interleukin 4 induced 1 | 16 | 191,143 | 194,187 | 3045 |
| 33 | TLR4 | Toll-like receptor 4 precursor | 17 | 3,566,454 | 3,571,907 | 5454 |
| 33 | BRINP1 | Bmp/retinoic acid inducible neural specific 1 | 17 | 3,985,882 | 4,064,290 | 78,409 |
| 33 | CD7 | T-cell antigen CD7 | 18 | 3,681,786 | 3,686,083 | 4298 |
| 35 | TAGLN | Transgelin | 24 | 5,229,236 | 5,232,028 | 2793 |
| 36 | TREML2 | Triggering receptor expressed on myeloid cells isoform 1 | 26 | 4,693,285 | 4,699,278 | 5994 |
| 37 | CD79B | CD79b molecule | 27 | 1,622,587 | 1,627,022 | 4436 |
| QTLR | Gene Name | Ensembl Gene ID | Chr | Start | End | Length |
|---|---|---|---|---|---|---|
| 5 | gga-mir-6553 | ENSGALG00000026406 | 1 | 76,381,420 | 76,381,519 | 100 |
| 7 | gga-mir-1738 | ENSGALG00000025522 | 1 | 104,085,133 | 104,085,223 | 91 |
| 12 | gga-mir-1749 | ENSGALG00000025364 | 2 | 569,326 | 569,423 | 98 |
| 12 | gga-mir-6710 | ENSGALG00000027384 | 2 | 724,388 | 724,497 | 110 |
| 18 | gga-mir-1462 | ENSGALG00000025207 | 4 | 8,488,386 | 8,488,495 | 110 |
| 25 | gga-mir-1659 | ENSGALG00000025154 | 7 | 13,177,026 | 13,177,126 | 101 |
| 25 | gga-mir-6624 | ENSGALG00000026751 | 7 | 15,758,919 | 15,759,028 | 110 |
| 25 | gga-mir-1570 | ENSGALG00000025238 | 7 | 16,274,218 | 16,274,317 | 100 |
| 26 | gga-mir-6660 | ENSGALG00000028738 | 10 | 1,076,742 | 1,076,851 | 110 |
| 30 | gga-mir-1644 | ENSGALG00000025220 | 14 | 8,124,516 | 8,124,601 | 86 |
| 31 | gga-mir-6643 | ENSGALG00000027935 | 14 | 13,143,450 | 13,143,559 | 110 |
| 31 | gga-mir-1588 | ENSGALG00000025419 | 14 | 14,125,510 | 14,125,611 | 102 |
| 31 | gga-mir-1715 | ENSGALG00000025576 | 14 | 14,285,835 | 14,285,936 | 102 |
| 35 | gga-mir-6612 | ENSGALG00000026265 | 24 | 4,779,927 | 4,780,036 | 110 |
| 35 | gga-mir-6671 | ENSGALG00000027111 | 24 | 5,035,783 | 5,035,892 | 110 |
| 35 | gga-mir-1745 | ENSGALG00000026550 | 24 | 5,163,387 | 5,163,473 | 87 |
| 37 | gga-mir-6573 | ENSGALG00000028331 | 27 | 1,532,553 | 1,532,671 | 119 |
| QTLR | Gene ID; Transcript ID; Stable Transcript ID | Chr | Start | End | Length | Distance | Strand | Exons |
|---|---|---|---|---|---|---|---|---|
| 1 | R192;R192.1;RI_1_8950428_8950983_1_1 | 1 | 8,950,428 | 8,950,983 | 556 | + | 1 | |
| 3 | R1291;R1291.1;RI_1_52122457_52122949_1_1 | 1 | 52,122,457 | 52,122,949 | 493 | 43,171,475 | − | 1 |
| 3 | R1292;R1292.1;RI_1_52183105_52322961_6_1 | 1 | 52,183,105 | 52,322,961 | 139,857 | 60,157 | − | 6 |
| 3 | R1293;R1293.2;RI_1_52481252_52482976_1_1 | 1 | 52,481,252 | 52,482,976 | 1725 | 158,292 | − | 1 |
| 3 | R1293;R1293.3;RI_1_52481282_52488508_2_1 | 1 | 52,481,282 | 52,488,508 | 7227 | −1693 | − | 2 |
| 20 | R12773;R12773.1;RI_5_7604020_7604920_1_1 | 5 | 7,604,020 | 7,604,920 | 901 | + | 1 | |
| 20 | R12779;R12779.1;RI_5_7727117_7728010_1_1 | 5 | 7,727,117 | 7,728,010 | 894 | 122,198 | − | 1 |
| 20 | R12784;R12784.1;RI_5_7960049_7961460_1_1 | 5 | 7,960,049 | 7,961,460 | 1412 | 232,040 | + | 1 |
| 21 | R13147;R13147.6;RI_5_18814123_18815319_2_1 | 5 | 18,814,123 | 18,815,319 | 1197 | 10,852,664 | + | 2 |
| 21 | R13148;R13148.1;RI_5_18861679_18862530_1_1 | 5 | 18,861,679 | 18,862,530 | 852 | 46,361 | + | 1 |
| 21 | R13149;R13149.1;RI_5_19632618_19793846_5_1 | 5 | 19,632,618 | 19,793,846 | 161,229 | 770,089 | − | 5 |
| 24 | R15316;R15316.1;RI_6_31138889_31157672_3_1 | 6 | 31,138,889 | 31,157,672 | 18,784 | − | 3 | |
| 24 | R15317;R15317.1;RI_6_31199730_31200561_1_1 | 6 | 31,199,730 | 31,200,561 | 832 | 42,059 | − | 1 |
| 24 | R15318;R15318.1;RI_6_31204510_31210163_7_1 | 6 | 31,204,510 | 31,210,163 | 5654 | 3950 | + | 7 |
| 24 | R15319;R15319.1;RI_6_31263768_31264516_1_1 | 6 | 31,263,768 | 31,264,516 | 749 | 53,606 | + | 1 |
| 24 | R15320;R15320.2;RI_6_31277294_31279639_1_1 | 6 | 31,277,294 | 31,279,639 | 2346 | 12,779 | − | 1 |
| 24 | R15321;R15321.1;RI_6_31368941_31369400_1_1 | 6 | 31,368,941 | 31,369,400 | 460 | 89,303 | − | 1 |
| QTLR | Gene | Marker | Chr | bp | Consequence | Effect |
|---|---|---|---|---|---|---|
| 14 | ENSGALG00000015219 | MD-07 | 2 | 107,385,545 | S/* | Stop gained |
| 25 | gga-mir-7474 | MD-16 | 7 | 14,743,048 | actgat | Frame shift |
| 26 | ENSGALG00000001269 | MD-20 | 10 | 48,846 | P/X | Frame shift |
| 29 | ENSGALG00000004078 | MD-23 | 13 | 11,619,922 | gtt/gGTTTtt | Frame shift |
| 30 | IGSF6 | MD-28 | 14 | 8,738,347 | L/X | Frame shift |
| 32 | BG1 | MD-29 | 16 | 117,493 | Splice site acceptor | |
| 32 | BG1 | MD-30 | 16 | 118,808 | Q/* | Stop gained |
| 32 | ENSGALG00000028367 | MD-32 | 16 | 206,100 | Splice site acceptor | |
| 32 | ENSGALG00000028367 | MD-33 | 16 | 206,241 | Splice site acceptor | |
| 36 | ENSGALG00000003188 | MD-44 | 26 | 4,629,538 | *C | Stop lost |
| 37 | SCN4A | MD-45 | 27 | 1,646,318 | aga/agaCAGAAAT | Frame shift |
| 37 | SCN4A | MD-46 | 27 | 1,646,601 | Q/* | Stop gained |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, J.; Lipkin, E.; Soller, M.; Fulton, J.E.; Burt, D.W. Mapping QTL Associated with Resistance to Avian Oncogenic Marek’s Disease Virus (MDV) Reveals Major Candidate Genes and Variants. Genes 2020, 11, 1019. https://doi.org/10.3390/genes11091019
Smith J, Lipkin E, Soller M, Fulton JE, Burt DW. Mapping QTL Associated with Resistance to Avian Oncogenic Marek’s Disease Virus (MDV) Reveals Major Candidate Genes and Variants. Genes. 2020; 11(9):1019. https://doi.org/10.3390/genes11091019
Chicago/Turabian StyleSmith, Jacqueline, Ehud Lipkin, Morris Soller, Janet E. Fulton, and David W. Burt. 2020. "Mapping QTL Associated with Resistance to Avian Oncogenic Marek’s Disease Virus (MDV) Reveals Major Candidate Genes and Variants" Genes 11, no. 9: 1019. https://doi.org/10.3390/genes11091019
APA StyleSmith, J., Lipkin, E., Soller, M., Fulton, J. E., & Burt, D. W. (2020). Mapping QTL Associated with Resistance to Avian Oncogenic Marek’s Disease Virus (MDV) Reveals Major Candidate Genes and Variants. Genes, 11(9), 1019. https://doi.org/10.3390/genes11091019