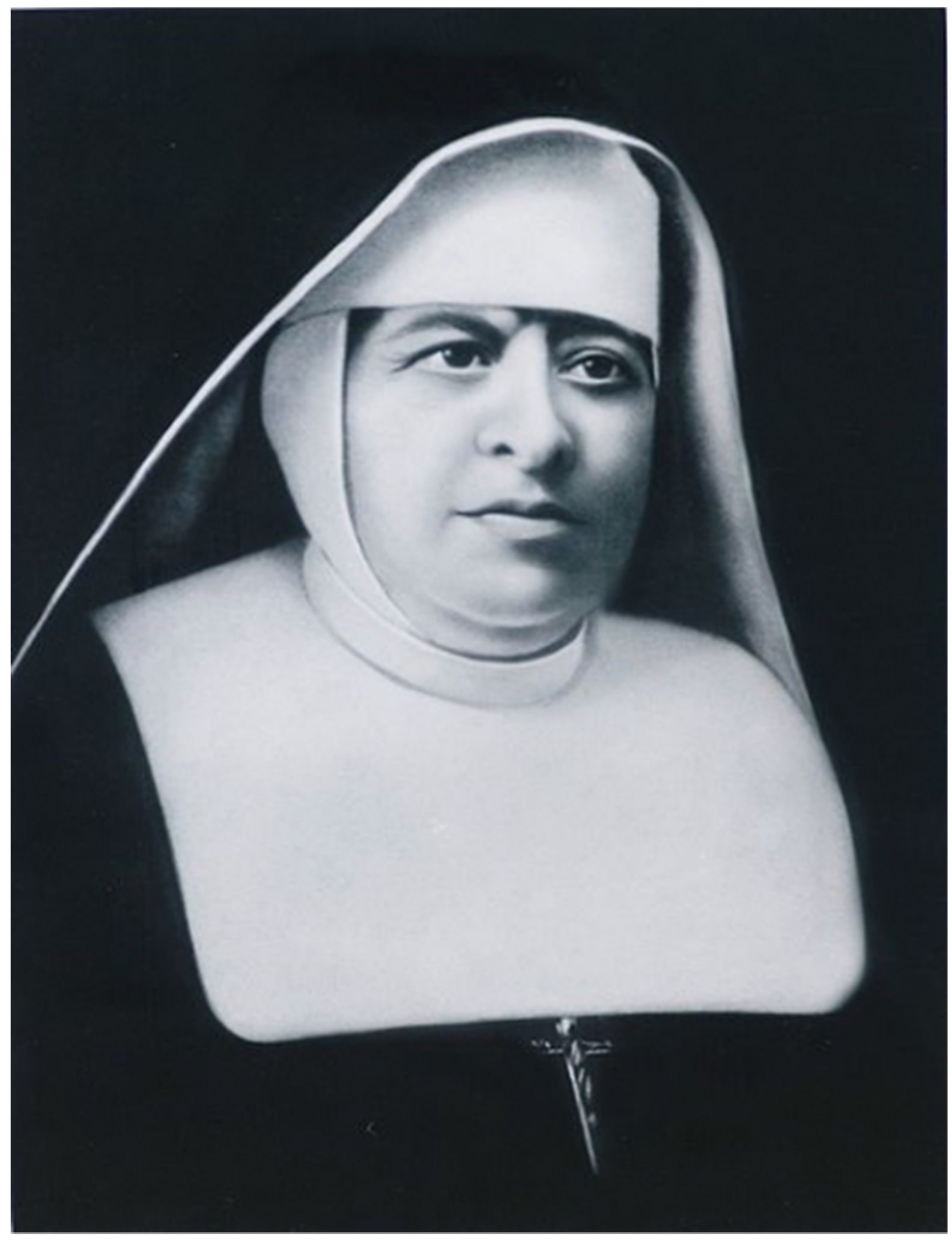
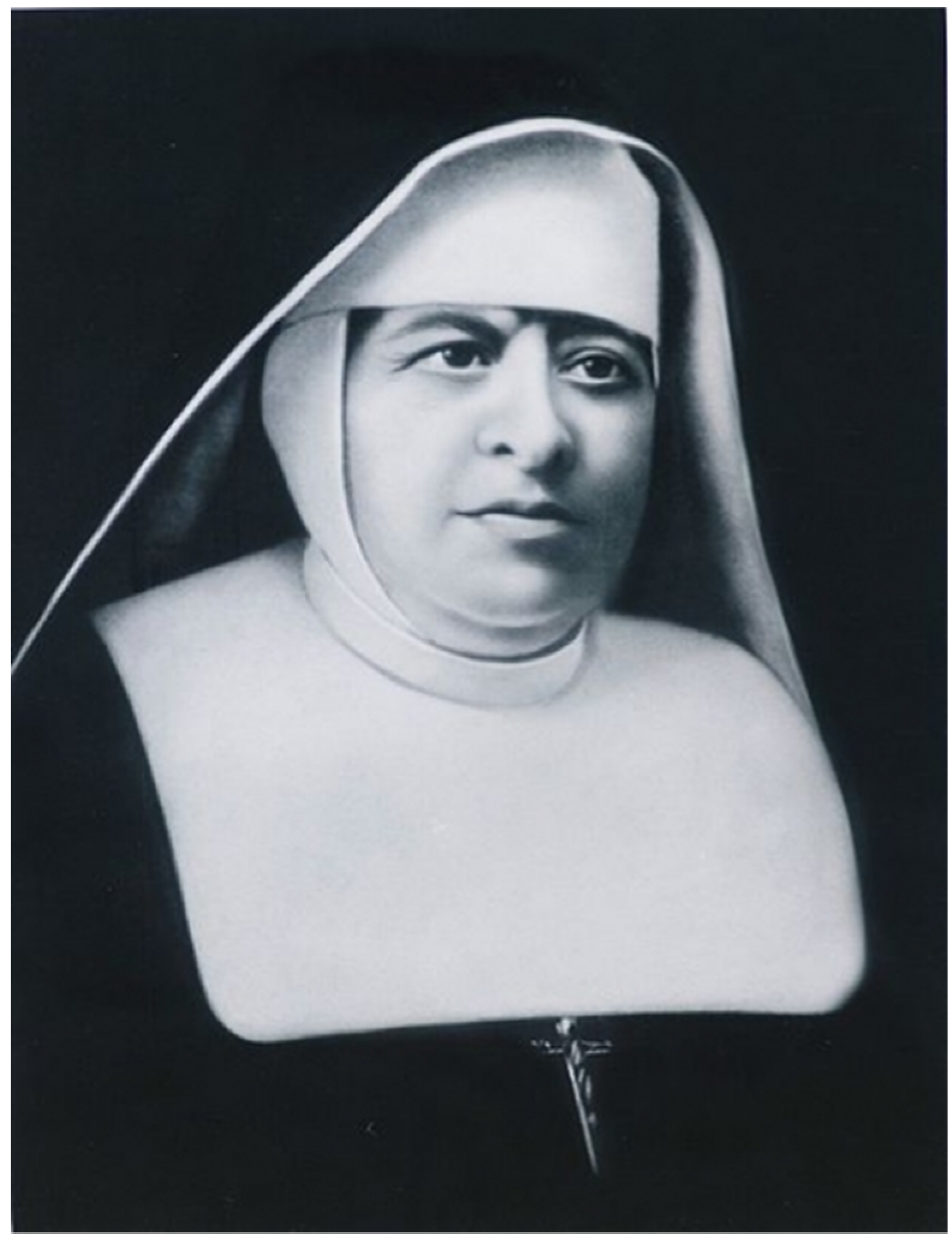
A Forensic Genomics Approach for the Identification of Sister Marija Crucifiksa Kozulić

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Selection
2.1.1. Skeletal Samples
2.1.2. DNA Reference Sample
2.2. DNA Extraction
2.2.1. Skeletal Samples
2.2.2. Buccal Swab
2.3. DNA Quantitation
2.4. Mitochondrial Genome Sequencing
2.5. Mitogenome Sequence Analysis
2.6. Autosomal STR and Identity SNP Sequencing
2.7. Autosomal STR and Identity SNP Data Analysis
2.8. Kinship Inference
3. Results
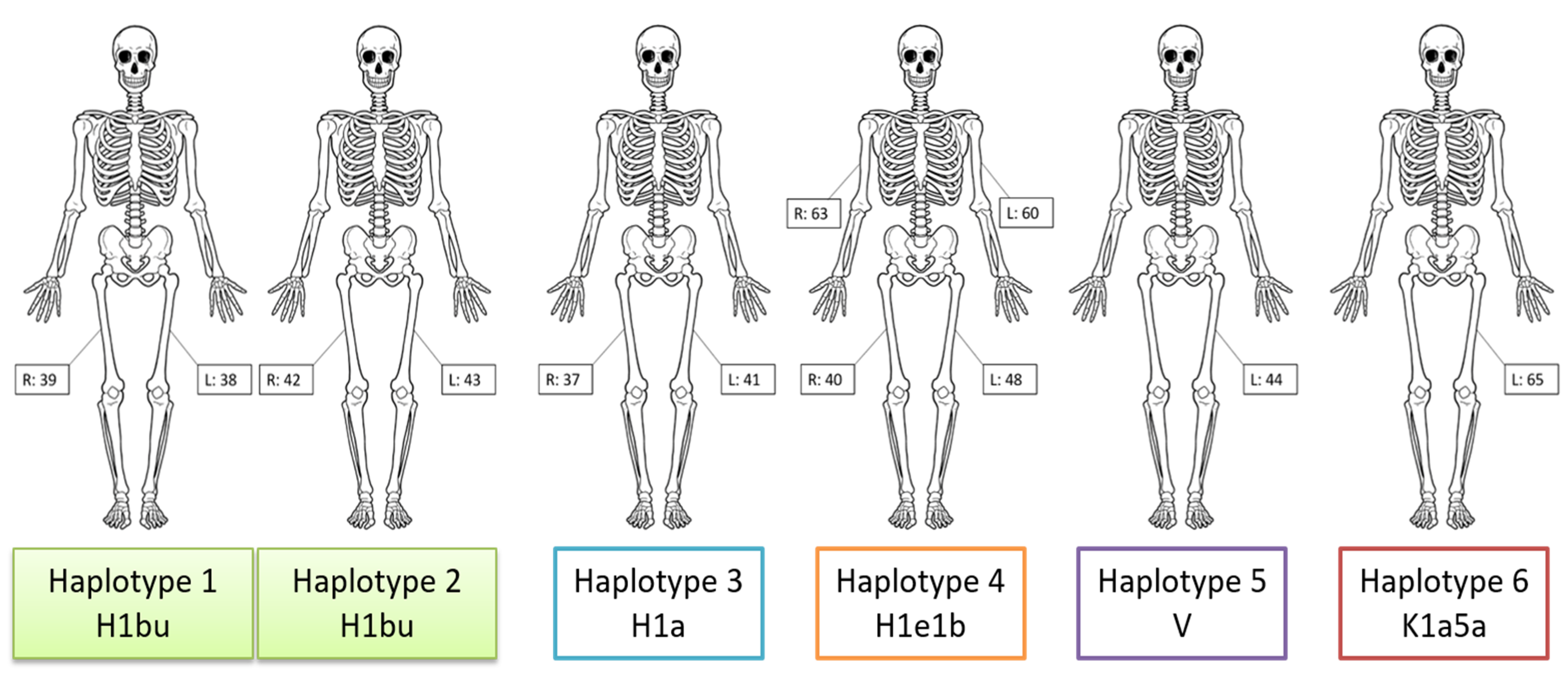
3.1. Mitogenome Sequencing
3.2. Autosomal STR and SNP Sequencing
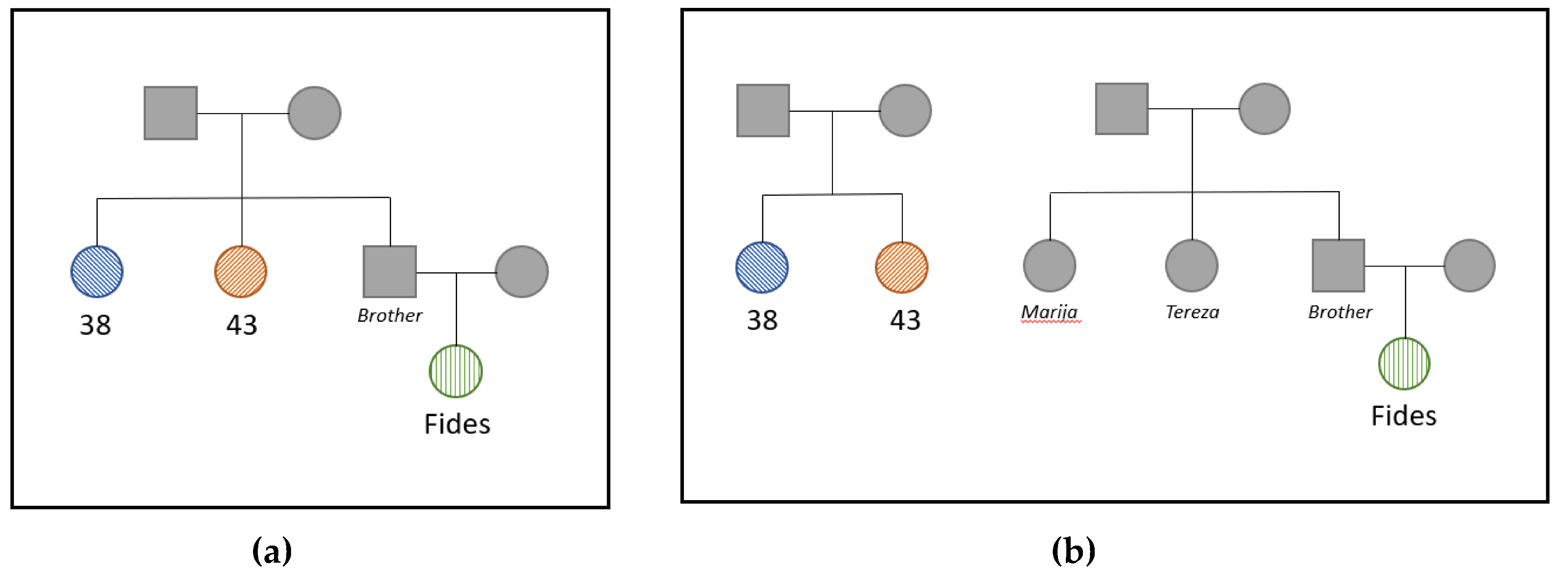
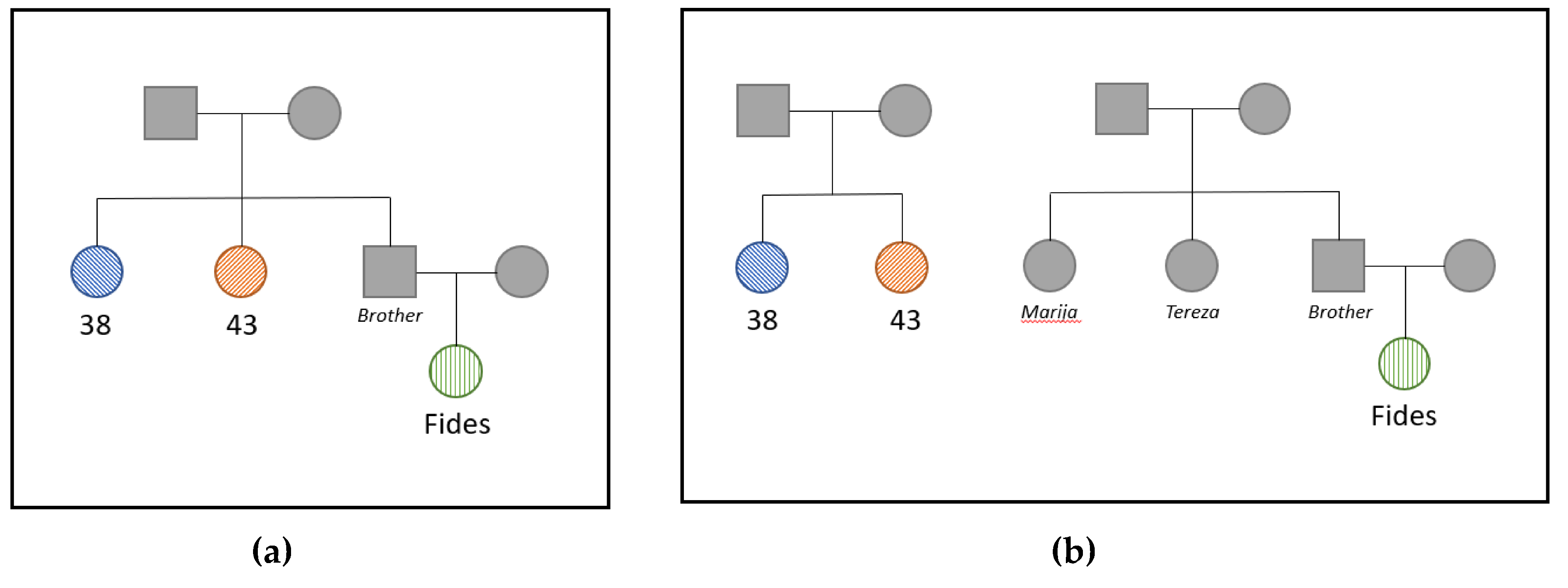
3.3. Kinship Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Mlakić, D. Mother Marija Krucifiksa Kozulić in Risika (In Croatian). In Družba Sestara Presvetog Srca Isusova: Postulatura Službenice Božje Majke Marije Krucifikse Kozuli; Denona: Zagreb, Croatia, 2017. [Google Scholar]
- Daniels-Higginbotham, J.; Gorden, E.M.; Farmer, S.K.; Spatola, B.; Damann, F.; Bellantoni, N.; Gagnon, K.S.; de la Puente, M.; Xavier, C.; Walsh, S. DNA Testing Reveals the Putative Identity of JB55, a 19th Century Vampire Buried in Griswold, Connecticut. Genes 2019, 10, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, C.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Oliver, R.S.; Barritt-Ross, S.; McMahon, T.P. Performance Evaluation of a Mitogenome Capture and Illumina Sequencing Protocol using Non-Probative, Case-Type Skeletal Samples: Implications for the use of a Positive Control in a Next-Generation Sequencing Procedure. Forensic Sci. Int. Genet. 2017, 31, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorden, E.M.; Sturk-Andreaggi, K.; Marshall, C. Repair of DNA Damage Caused by Cytosine Deamination in Mitochondrial DNA of Forensic Case Samples. Forensic Sci. Int. Genet. 2018, 34, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallimore, J.M.; McElhoe, J.A.; Holland, M.M. Assessing Heteroplasmic Variant Drift in the mtDNA Control Region of Human Hairs using an MPS Approach. Forensic Sci. Int. Genet. 2018, 32, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and Revision of the Cambridge Reference Sequence for Human Mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Sturk-Andreaggi, K.; Peck, M.A.; Boysen, C.; Dekker, P.; McMahon, T.P.; Marshall, C.K. AQME: A Forensic Mitochondrial DNA Analysis Tool for Next-Generation Sequencing Data. Forensic Sci. Int. Genet. 2017, 31, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.M.; Pack, E.D.; McElhoe, J.A. Evaluation of GeneMarker® HTS for Improved Alignment of mtDNA MPS Data, Haplotype Determination, and Heteroplasmy Assessment. Forensic Sci. Int. Genet. 2017, 28, 90–98. [Google Scholar] [CrossRef]
- Gill, P.; Whitaker, J.; Flaxman, C.; Brown, N.; Buckleton, J. An Investigation of the Rigor of Interpretation Rules for STRs Derived from Less than 100 Pg of DNA. Forensic Sci. Int. 2000, 112, 17–40. [Google Scholar] [CrossRef]
- Whitaker, J.P.; Cotton, E.A.; Gill, P. A Comparison of the Characteristics of Profiles Produced with the AMPFlSTR SGM Plus Multiplex System for both Standard and Low Copy Number (LCN) STR DNA Analysis. Forensic Sci. Int. 2001, 123, 215–223. [Google Scholar] [CrossRef]
- Egeland, T.; Mostad, P.F.; Mevag, B.; Stenersen, M. Beyond Traditional Paternity and Identification Cases. Selecting the most Probable Pedigree. Forensic Sci. Int. 2000, 110, 47–59. [Google Scholar] [CrossRef]
- Kling, D.; Tillmar, A.O.; Egeland, T. Familias 3–Extensions and New Functionality. Forensic Sci. Int. Genet. 2014, 13, 121–127. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Durbin, R.M.; Abecasis, G.R.; Altshuler, D.L.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A Map of Human Genome Variation from Population-Scale Sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 1000 Genomes Project Consortium. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, L.; Jin, Y.; Zhang, H.; Qiang, W.; Shekhtman, E.; Shao, D.; Revoe, D.; Villamarin, R.; Ivanchenko, E.; Kimura, M.; et al. ALFA: Allele Frequency Aggregator. National Center for Biotechnology Information, U.S. National Library of Medicine. Available online: www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/ (accessed on 10 March 2020).
- Steffen, C.R.; Coble, M.D.; Gettings, K.B.; Vallone, P.M. Corrigendum to ‘;U.S. Population Data for 29 Autosomal STR Loci’ [Forensic Sci. Int. Genet. 7 (2013) e82-e83]. Forensic Sci. Int. Genet. 2017, 31, e36–e40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruitberg, C.M.; Reeder, D.J.; Butler, J.M. STRBase: A Short Tandem Repeat DNA Database for the Human Identity Testing Community. Nucleic Acids Res. 2001, 29, 320–322. [Google Scholar] [CrossRef]
- Gjertson, D.W.; Brenner, C.H.; Baur, M.P.; Carracedo, A.; Guidet, F.; Luque, J.A.; Lessig, R.; Mayr, W.R.; Pascali, V.L.; Prinz, M.; et al. ISFG: Recommendations on Biostatistics in Paternity Testing. Forensic Sci. Int. Genet. 2007, 1, 223–231. [Google Scholar] [CrossRef]
- Holland, M.; Makova, K.; McElhoe, J. Deep-Coverage MPS Analysis of Heteroplasmic Variants within the mtGenome Allows for Frequent Differentiation of Maternal Relatives. Genes 2018, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Parson, W.; Gusmao, L.; Hares, D.; Irwin, J.; Mayr, W.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P. DNA Commission of the International Society for Forensic Genetics: Revised and Extended Guidelines for Mitochondrial DNA Typing. Forensic Sci. Int. Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef]
- Ma, K.; Zhao, X.; Li, H.; Cao, Y.; Li, W.; Ouyang, J.; Xie, L.; Liu, W. Massive Parallel Sequencing of Mitochondrial DNA Genomes from Mother-Child Pairs using the Ion Torrent Personal Genome Machine (PGM). Forensic Sci. Int. Genet. 2018, 32, 88–93. [Google Scholar] [CrossRef]
- Rebolledo-Jaramillo, B.; Su, M.S.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal Age Effect and Severe Germ-Line Bottleneck in the Inheritance of Human Mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479. [Google Scholar] [CrossRef] [Green Version]
- Parsons, T.J.; Huel, R.M.; Bajunović, Z.; Rizvić, A. Large Scale DNA Identification: The ICMP Experience. Forensic Sci. Int. Genet. 2019, 38, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Zavala, E.I.; Rajagopal, S.; Perry, G.H.; Kruzic, I.; Bašić, Ž; Parsons, T.J.; Holland, M.M. Impact of DNA Degradation on Massively Parallel Sequencing-Based Autosomal STR, iiSNP, and Mitochondrial DNA Typing Systems. Int. J. Leg. Med. 2019, 133, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Andelinovic, S.; Martin, P.; Sutlovic, D.; Erceg, I.; Huffine, E.; de Simon, L.F.; Albarran, C.; Definis-Gojanovic, M.; Fernandez-Rodriguez, A.; et al. DNA Typing from Skeletal Remains: Evaluation of Multiplex and Megaplex STR Systems on DNA Isolated from Bone and Teeth Samples. Croat. Med. J. 2001, 42, 260–266. [Google Scholar] [PubMed]

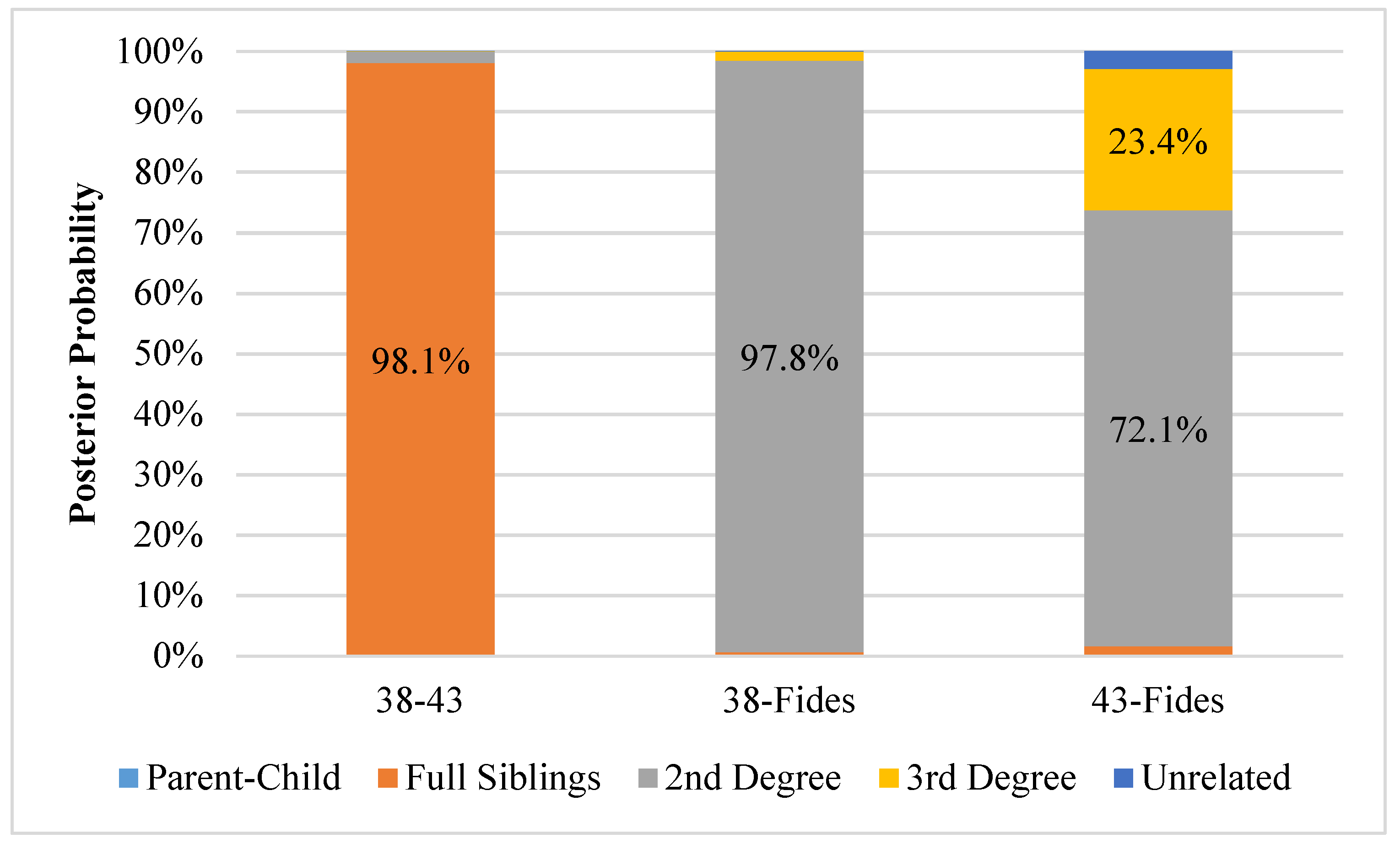

{kind=link}
{kind=link}
{kind=link}
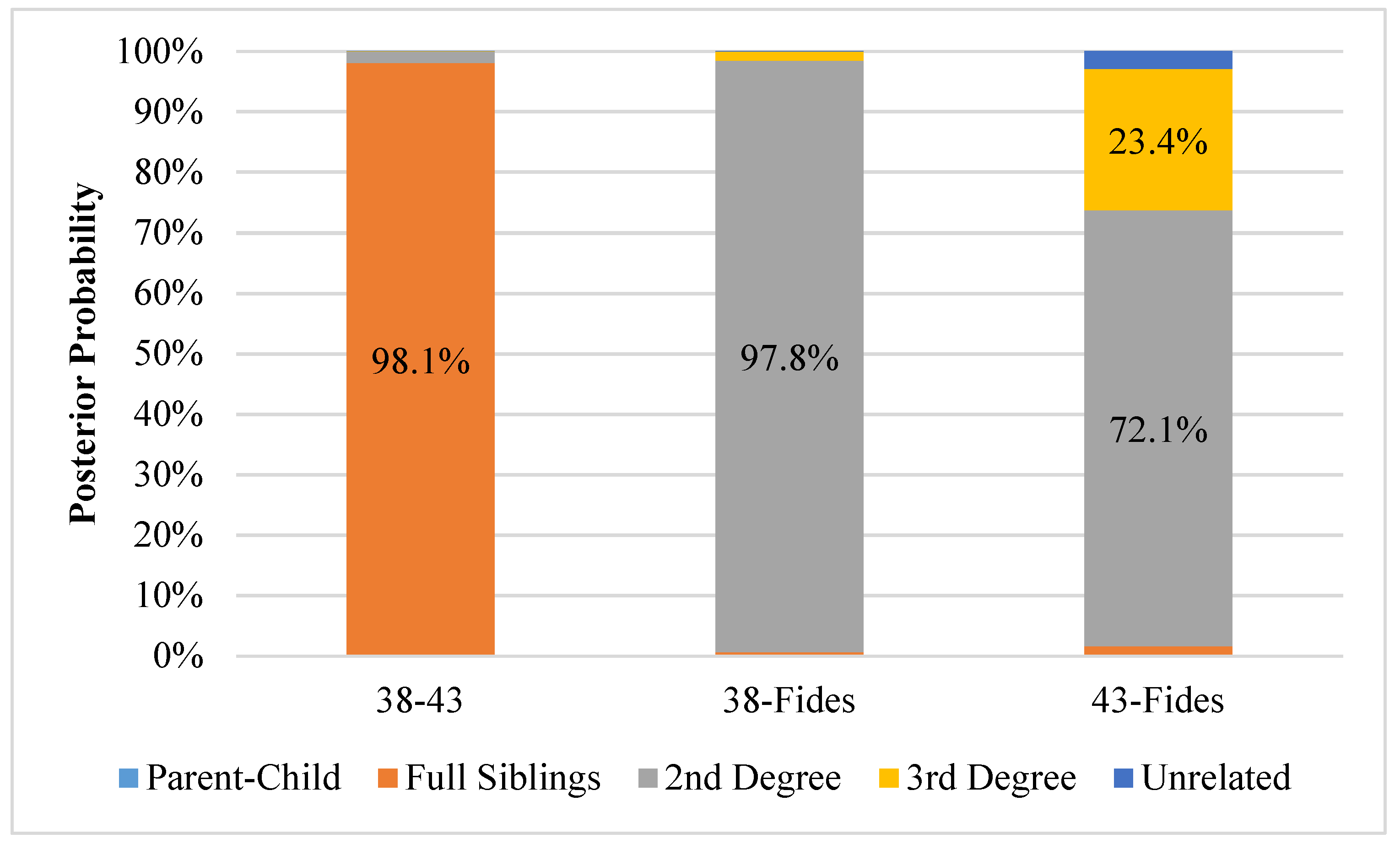{kind=link}
{kind=link}
| Sample ID | Reads | % Reads Mapped to rCRS | Reads Mapped to rCRS | Unique Reads Mapped to rCRS | Mean Mapped Read Length | Average Read Death | Bases ≥ 10X |
|---|---|---|---|---|---|---|---|
| 37.1 | 673,054 | 10.72% | 72,174 | 68,242 | 94.88 | 183.1 | 16,569 |
| 37.2 | 798,954 | 8.91% | 71,213 | 66,379 | 97.33 | 182.7 | 16,569 |
| 38.1 | 9,408,738 | 65.47% | 6,159,871 | 2,711,409 | 120.98 | 9557.6 | 16,569 |
| 38.2 | 8,937,744 | 45.81% | 4,094,273 | 2,163,767 | 134.69 | 8578.4 | 16,569 |
| 38.3 | 18,372,080 | 2.10% | 386,546 | 306,340 | 95.39 | 827.2 | 16,569 |
| 39.1 | 1,865,914 | 45.69% | 852,586 | 713,852 | 130.54 | 2717.3 | 16,569 |
| 39.2 | 8,216,798 | 16.20% | 1,331,479 | 1,024,117 | 130.95 | 3872 | 16,569 |
| 40.1 | 8,734,042 | 76.51% | 6,682,424 | 4,216,710 | 111.65 | 13,641.1 | 16,569 |
| 40.2 | 4,369,510 | 72.15% | 3,152,582 | 2,214,763 | 107.61 | 6846.9 | 16,569 |
| 40.3 | 6,759,524 | 55.25% | 3,734,350 | 2,370,261 | 96.16 | 6689.7 | 16,569 |
| 41.1 | 3,305,366 | 48.98% | 1,618,905 | 1,416,205 | 119.74 | 4790.5 | 16,569 |
| 41.2 | 2,818,352 | 49.54% | 1,396,161 | 1,238,362 | 119.16 | 4147.6 | 16,569 |
| 42.1 | 5,451,990 | 81.18% | 4,425,748 | 3,065,788 | 107.65 | 9555.1 | 16,569 |
| 42.2 | 9,205,980 | 80.18% | 7,381,722 | 4,674,287 | 105.68 | 14,355.1 | 16,569 |
| 43.1 | 2,596,506 | 43.95% | 1,141,189 | 949,031 | 98.46 | 2643.8 | 16,569 |
| 43.2 | 2,856,978 | 34.62% | 989,133 | 832,220 | 96.86 | 2285.1 | 16,569 |
| 43.3 | 14,864,996 | 70.57% | 10,489,877 | 4,013,492 | 86.22 | 10,229.2 | 16,569 |
| 44.1 | 1,751,348 | 1.02% | 17,928 | 17,525 | 102.58 | 48 | 16,569 |
| 44.2 | 2,184,922 | 1.84% | 40,259 | 39,103 | 99.71 | 104.4 | 16,569 |
| 45.1 | 1,718,530 | 1.95% | 33,482 | 30,812 | 117.08 | 102.2 | 16,569 |
| 45.2 | 116,968 | 1.67% | 1953 | 1877 | 87.91 | 4.6 | 1376 |
| 46.1 | 1,799,746 | 0.01% | 195 | 191 | 102.53 | 0.2 | 0 |
| 46.2 | 891,934 | 0.03% | 226 | 217 | 104.73 | 0.3 | 0 |
| 48.1 | 6,485,124 | 76.42% | 4,955,891 | 3,167,070 | 123.58 | 11,315.9 | 16,569 |
| 48.2 | 11,188,962 | 72.89% | 8,155,496 | 4,684,708 | 126.54 | 17,251.5 | 16,569 |
| 60.1 | 2,339,920 | 40.06% | 937,401 | 709,827 | 114.12 | 2367.6 | 16,569 |
| 60.2 | 1,692,924 | 41.48% | 702,247 | 533,893 | 117.25 | 1823.8 | 16,569 |
| 63.1 | 873,150 | 12.30% | 107,368 | 100,670 | 116.93 | 332.2 | 16,569 |
| 63.2 | 940,844 | 12.53% | 117,848 | 109,408 | 119.97 | 371.8 | 16,569 |
| 65.1 | 815,424 | 46.94% | 382,728 | 341,506 | 115.76 | 1148.5 | 16,569 |
| 65.2 | 222,180 | 5.62% | 12,479 | 12,081 | 139.45 | 47.6 | 16,569 |
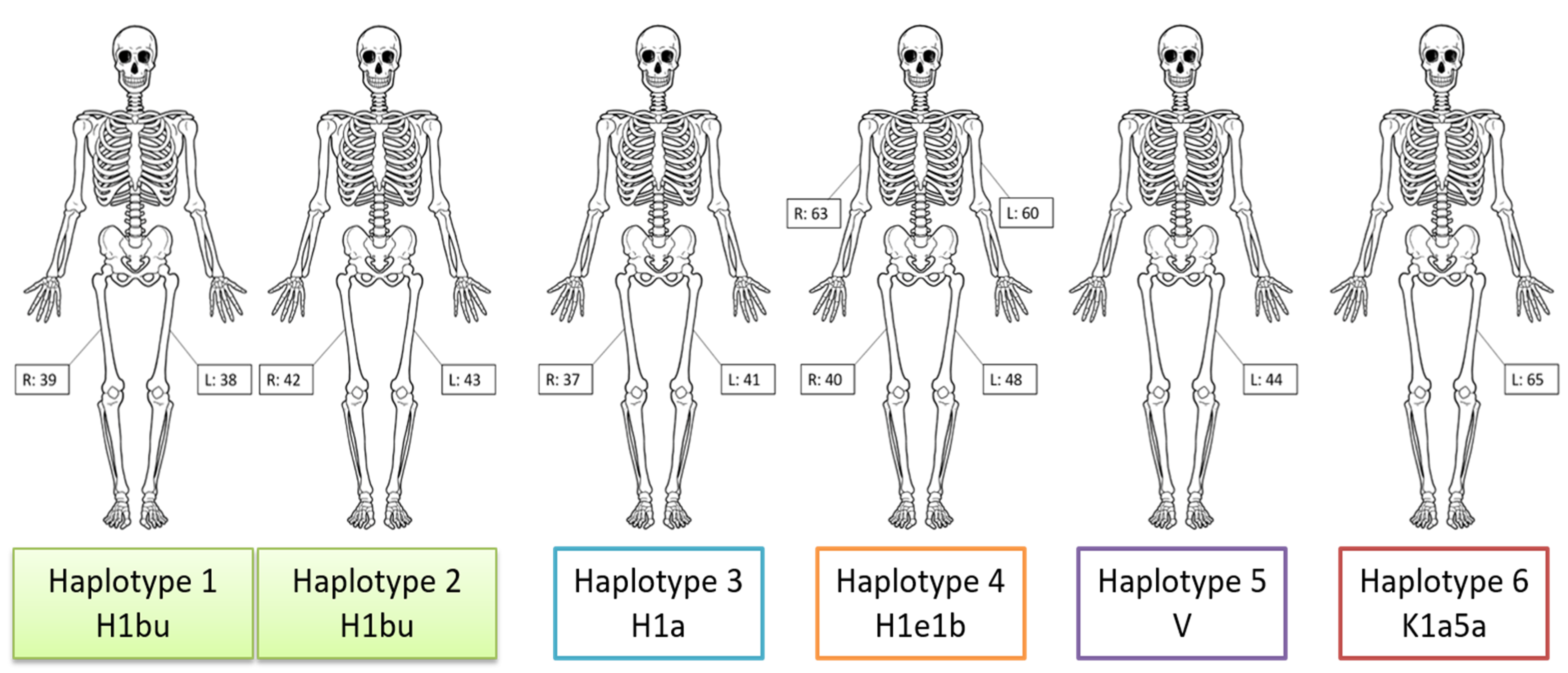
| Sample(s) | Haplogroup | Haplotype |
|---|---|---|
| 38, 39 | H1bu | 263G 309.1C 315.1C 750G 1438G 3010A 4769G 5558G 8860G 12337.1c 13327R 15326G 16519C |
| 42, 43 | H1bu | 263G 309.1C 315.1C 750G 1438G 3010A 4769G 5558G 8860G 13327R 15326G 16519C |
| 37, 41 | H1a | 73G 263G 309.1C 315.1C 750G 1438G 3010A 4769G 8860G 15326G 16162G 16519C |
| 40, 48, 60, 63 | H1e1b | 263G 309.1C 315.1C 453C 750G 1438G 3010A 4769G 5460A 8512G 8860G 10274C 15326G 16519C |
| 44 | V | 72C 263G 315.1C 750G 1327R 1438G 2706G 4580A 4769G 7028T 8860G 12408C 15326G 15904T 16298C |
| 65 | K1a5a | 73G 263G 315.1C 497T 524.1A 524.2C 750G 1189C 1438G 1811G 2706G 3480G 4640T 4769G 7028T 8860G 9055A 9647C 9698C 10398G 10550G 11017C 11299C 11467G 11719A 12308G 12372A 14167T 14766T 14798C 15326G 16093Y 16129A 16224C 16311C 16362C 16519C |
| Sample | AuSTRs (n = 29) | SNPs (n = 90) | Total Loci (n = 119) |
|---|---|---|---|
| 38 | 22 (76%) | 67 (74%) | 89 (75%) |
| 40 | 29 (100%) | 71 (79%) | 100 (84%) |
| 43 | 4 (14%) | 38 (42%) | 42 (35%) |
| Buccal | 27 (93%) | 85 (94%) | 112 (94%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marshall, C.; Sturk-Andreaggi, K.; Gorden, E.M.; Daniels-Higginbotham, J.; Sanchez, S.G.; Bašić, Ž.; Kružić, I.; Anđelinović, Š.; Bosnar, A.; Čoklo, M.; et al. A Forensic Genomics Approach for the Identification of Sister Marija Crucifiksa Kozulić. Genes 2020, 11, 938. https://doi.org/10.3390/genes11080938
Marshall C, Sturk-Andreaggi K, Gorden EM, Daniels-Higginbotham J, Sanchez SG, Bašić Ž, Kružić I, Anđelinović Š, Bosnar A, Čoklo M, et al. A Forensic Genomics Approach for the Identification of Sister Marija Crucifiksa Kozulić. Genes. 2020; 11(8):938. https://doi.org/10.3390/genes11080938
Chicago/Turabian StyleMarshall, Charla, Kimberly Sturk-Andreaggi, Erin M. Gorden, Jennifer Daniels-Higginbotham, Sidney Gaston Sanchez, Željana Bašić, Ivana Kružić, Šimun Anđelinović, Alan Bosnar, Miran Čoklo, and et al. 2020. "A Forensic Genomics Approach for the Identification of Sister Marija Crucifiksa Kozulić" Genes 11, no. 8: 938. https://doi.org/10.3390/genes11080938
APA StyleMarshall, C., Sturk-Andreaggi, K., Gorden, E. M., Daniels-Higginbotham, J., Sanchez, S. G., Bašić, Ž., Kružić, I., Anđelinović, Š., Bosnar, A., Čoklo, M., Petaros, A., McMahon, T. P., Primorac, D., & Holland, M. M. (2020). A Forensic Genomics Approach for the Identification of Sister Marija Crucifiksa Kozulić. Genes, 11(8), 938. https://doi.org/10.3390/genes11080938